scanpy预处理总结
欢迎关注我们组的微信公众号,更多好文章在等你呦!
微信公众号名:碳硅数据
公众号二维码:

记录一下关于scanpy preprocessing的结果
import scanpy as sc
adata = sc.read("/Users/yxk/Desktop/test_dataset/pbmc/pbmc.h5ad")
print(adata)


可以看到这个原始数据是稀疏矩阵形式的,
如果经过预处理
sc.pp.normalize_total(adata,target_sum=1e4)
sc.pp.log1p(adata)
sc.pp.highly_variable_genes(adata,n_top_genes=2000,subset=True)
sc.pp.scale(adata)
sc.tl.pca(adata)
结果如下

这个还很正常,
但是我今天测试了别人的一个预处理代码
import torch
import torch
import torch.nn as nn
import numpy as np
import sys
import random
import os
from torch.utils.data import DataLoader
from tqdm import tqdm
import pandas as pd
import scipy
from scipy.sparse import issparse, csr
from anndata import AnnData
import scanpy as sc
from sklearn.preprocessing import maxabs_scale, MaxAbsScaler
CHUNK_SIZE = 20000
##### set random seed for reproduce result #####
def seed_torch(seed=1029):
random.seed(seed)
os.environ['PYTHONHASHSEED'] = str(seed)
np.random.seed(seed)
torch.manual_seed(seed)
torch.cuda.manual_seed(seed)
torch.cuda.manual_seed_all(seed) # if you are using multi-GPU.
torch.backends.cudnn.badatahmark = False
torch.backends.cudnn.deterministic = True
torch.backends.cudnn.enabled = False
def predict(model,X,batch_size=128):
"""
prediction for data matrix(produce embedding)
Argument:
------------------------------------------------------------------
X: data matrix fo dataset
batch_size: batch_size for dataloader
------------------------------------------------------------------
"""
device=torch.device("cpu")
dataloader = DataLoader(
torch.FloatTensor(X), batch_size=batch_size, pin_memory=False, shuffle=False
)
data_iterator = tqdm(dataloader, leave=False, unit="batch")
model=model.to(device)
with torch.no_grad():
model.eval()
features = []
recons = []
for batch in data_iterator:
batch = batch.to(device)
output,recon = model(batch)
features.append(
output.detach().cpu()
) # move to the CPU to prevent out of memory on the GPU
recons.append(
recon.detach().cpu()
)
features=torch.cat(features).cpu().numpy()
recons = torch.cat(recons).cpu().numpy()
return features,recons
# This will be used to create train and val sets
class BasicDataset(torch.utils.data.Dataset):
def __init__(self, data, labels):
self.data = torch.from_numpy(data).float()
self.labels = labels
def __getitem__(self, index):
return self.data[index], self.labels[index]
def __len__(self):
return len(self.data)
def preprocessing_rna(
adata: AnnData,
min_features: int = 600,
min_cells: int = 3,
target_sum: int = 10000,
n_top_features = 2000, # or gene list
chunk_size: int = CHUNK_SIZE,
backed: bool = False,
log=None
):
"""
Preprocessing single-cell RNA-seq data
Parameters
----------
adata
An AnnData matrice of shape n_obs x n_vars. Rows correspond to cells and columns to genes.
min_features
Filtered out cells that are detected in less than n genes. Default: 600.
min_cells
Filtered out genes that are detected in less than n cells. Default: 3.
target_sum
After normalization, each cell has a total count equal to target_sum. If None, total count of each cell equal to the median of total counts for cells before normalization.
n_top_features
Number of highly-variable genes to keep. Default: 2000.
chunk_size
Number of samples from the same batch to transform. Default: 20000.
log
If log, record each operation in the log file. Default: None.
Return
-------
The AnnData object after preprocessing.
"""
if min_features is None: min_features = 600
if n_top_features is None: n_top_features = 2000
if target_sum is None: target_sum = 10000
if log: log.info('Preprocessing')
# if not issparse(adata.X):
if type(adata.X) != csr.csr_matrix and (not backed) and (not adata.isbacked):
adata.X = scipy.sparse.csr_matrix(adata.X)
adata = adata[:, [gene for gene in adata.var_names
if not str(gene).startswith(tuple(['ERCC', 'MT-', 'mt-']))]]
if log: log.info('Filtering cells')
sc.pp.filter_cells(adata, min_genes=min_features)
if log: log.info('Filtering features')
sc.pp.filter_genes(adata, min_cells=min_cells)
if log: log.info('Normalizing total per cell')
sc.pp.normalize_total(adata, target_sum=target_sum)
if log: log.info('Log1p transforming')
sc.pp.log1p(adata)
adata.raw = adata
if log: log.info('Finding variable features')
if type(n_top_features) == int and n_top_features>0:
sc.pp.highly_variable_genes(adata, n_top_genes=n_top_features, batch_key='batch', inplace=False, subset=True)
elif type(n_top_features) != int:
adata = reindex(adata, n_top_features)
if log: log.info('Batch specific maxabs scaling')
# adata = batch_scale(adata, chunk_size=chunk_size)
adata.X = MaxAbsScaler().fit_transform(adata.X)
if log: log.info('Processed dataset shape: {}'.format(adata.shape))
return adata
def reindex(adata, genes, chunk_size=CHUNK_SIZE):
"""
Reindex AnnData with gene list
Parameters
----------
adata
AnnData
genes
gene list for indexing
chunk_size
chunk large data into small chunks
Return
------
AnnData
"""
idx = [i for i, g in enumerate(genes) if g in adata.var_names]
print('There are {} gene in selected genes'.format(len(idx)))
if len(idx) == len(genes):
adata = adata[:, genes]
else:
new_X = scipy.sparse.lil_matrix((adata.shape[0], len(genes)))
new_X[:, idx] = adata[:, genes[idx]].X
# for i in range(new_X.shape[0]//chunk_size+1):
# new_X[i*chunk_size:(i+1)*chunk_size, idx] = adata[i*chunk_size:(i+1)*chunk_size, genes[idx]].X
adata = AnnData(new_X.tocsr(), obs=adata.obs, var={'var_names':genes})
return adata
data_dir = '/Users/yxk/Desktop/test_dataset/pbmc/pbmc.h5ad'
adata = sc.read(data_dir)
adata.obs["BATCH"] = adata.obs["batch"].copy()
print(adata.X)
adata = preprocessing_rna(adata)
print(adata.X)
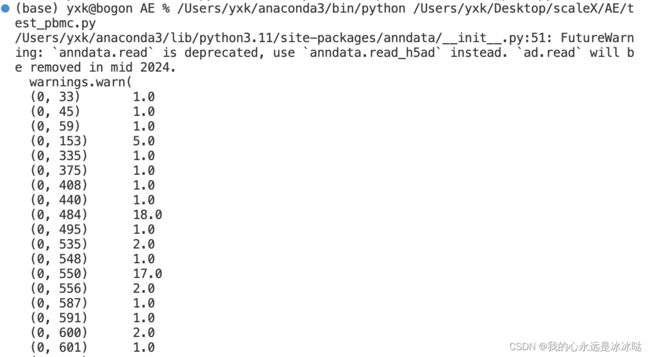

但是这个代码的预处理就是稀疏矩阵处理完后还是稀疏矩阵