scanpy 教程 1:预处理和聚类 3k PBMCs
「写在前面」
学习一个软件最好的方法就是啃它的官方文档。本着自己学习、分享他人的态度,分享官方文档的中文教程。软件可能随时更新,建议配合官方文档一起阅读。推荐先按顺序阅读往期内容:
文献篇:
1.文献阅读:SCANPY:大规模单细胞基因表达数据分析
2.文献阅读:scverse 项目为单细胞组学数据分析提供了计算生态系统
目录
- 1 预处理
- 2 主成分分析
- 3 计算邻域图
- 4 嵌入邻域图
- 5 对邻域图进行聚类
- 6 寻找标记基因
官网教程:
https://scanpy-tutorials.readthedocs.io/en/latest/pbmc3k.html
从 2017 年 5 月开始,Scanpy 允许复制 Seurat 的大部分引导聚类教程(Satija et al., 2015)。
我们非常感谢 Seurat 的作者提供的教程!与此同时,我们增加和删除了一些部分。
该数据由来自健康捐赠者的 3k PBMCs 组成,可从 10x Genomics 免费获得(here from this webpage)。在 unix 系统上,您可以取消注释并运行以下操作来下载和解包数据。最后一行创建了一个目录,用于写入已处理的数据。
mkdir data
wget http://cf.10xgenomics.com/samples/cell-exp/1.1.0/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz -O data/pbmc3k_filtered_gene_bc_matrices.tar.gz
cd data; tar -xzf pbmc3k_filtered_gene_bc_matrices.tar.gz
mkdir write
点击 GitHub 上的编辑按钮下载这个 notebook。在 GitHub 上,您可以通过右键单击并将链接另存为,使用 Raw 按钮进行下载。或者,下载整个 scanpy-tutorial repository。
在 Jupyter notebooks and lab 中,您可以通过点击 SHIFT+TAB 来查看 python 函数的文档。点击两次以展开视图。
import numpy as np
import pandas as pd
import scanpy as sc
sc.settings.verbosity = 3 # verbosity: errors (0), warnings (1), info (2), hints (3)
sc.logging.print_header()
sc.settings.set_figure_params(dpi=80, facecolor='white')
results_file = 'write/pbmc3k.h5ad' # the file that will store the analysis results
将计数矩阵读入 AnnData 对象,该对象包含许多用于注释和不同数据表示的 slots。它还自带基于 HDF5 的文件格式:.h5ad。
adata = sc.read_10x_mtx(
'data/filtered_gene_bc_matrices/hg19/', # the directory with the `.mtx` file
var_names='gene_symbols', # use gene symbols for the variable names (variables-axis index)
cache=True) # write a cache file for faster subsequent reading
有关 AnData 的更全面介绍,请参阅 anndata-tutorials/getting-started。
adata.var_names_make_unique() # this is unnecessary if using `var_names='gene_ids'` in `sc.read_10x_mtx`
adata
## AnnData object with n_obs × n_vars = 2700 × 32738
## var: 'gene_ids'
1 预处理
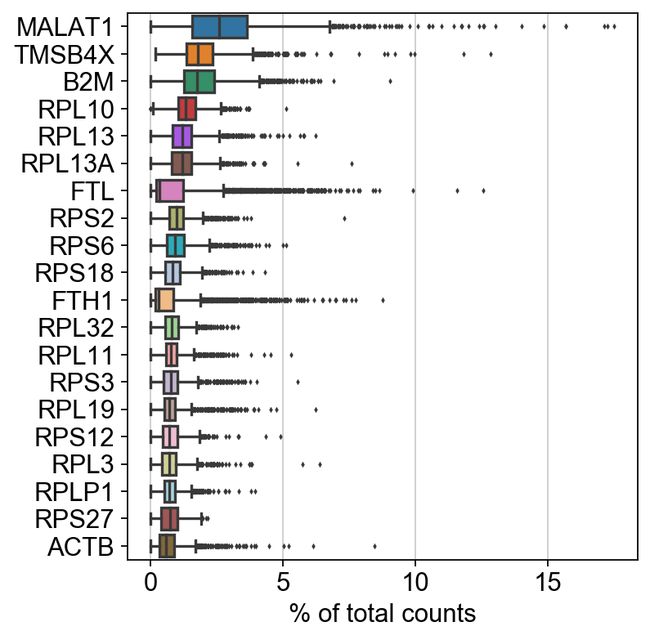
展示所有细胞中在每个细胞中高表达的基因。
sc.pl.highest_expr_genes(adata, n_top=20, )
基本过滤:
sc.pp.filter_cells(adata, min_genes=200)
sc.pp.filter_genes(adata, min_cells=3)
## filtered out 19024 genes that are detected in less than 3 cells
让我们收集一些关于线粒体基因的信息,这些信息对质量控制很重要。
Citing from “Simple Single Cell” workflows (Lun, McCarthy & Marioni, 2017):
高比例表明细胞质量差(Islam et al. 2014; Ilicic et al. 2016),可能是因为穿孔细胞的细胞质 RNA 丢失。其理由是线粒体比单个转录物分子大,不太可能通过细胞膜中的撕裂逃逸。
使用 pp.cacalculate_qc_metrics,我们可以非常高效地计算许多度量。
adata.var['mt'] = adata.var_names.str.startswith('MT-') # annotate the group of mitochondrial genes as 'mt'
sc.pp.calculate_qc_metrics(adata, qc_vars=['mt'], percent_top=None, log1p=False, inplace=True)
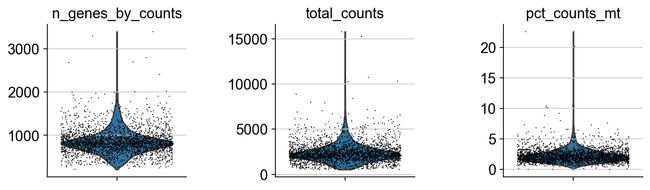
一些计算质量指标的小提琴图:
- 计数矩阵中表达的基因数量
- 每个细胞的总计数
- 线粒体基因计数的百分比
sc.pl.violin(adata, ['n_genes_by_counts', 'total_counts', 'pct_counts_mt'],
jitter=0.4, multi_panel=True)
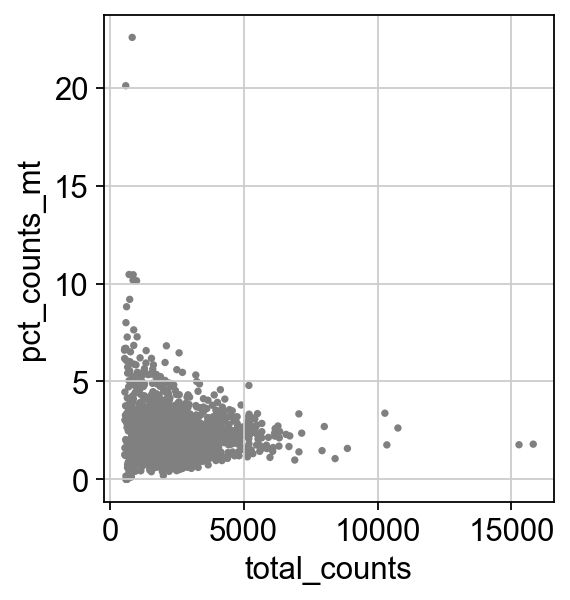
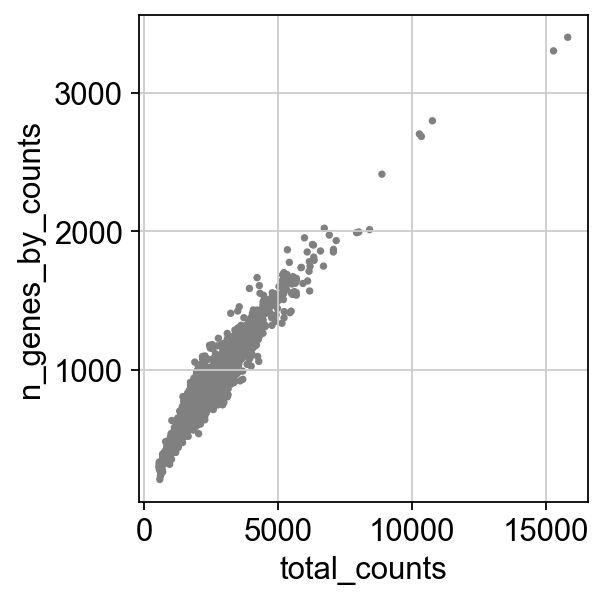
去除线粒体基因表达过多或总计数过多的细胞:
sc.pl.scatter(adata, x='total_counts', y='pct_counts_mt')
sc.pl.scatter(adata, x='total_counts', y='n_genes_by_counts')
实际上,通过对 AnData 对象进行切片来进行过滤。
adata = adata[adata.obs.n_genes_by_counts < 2500, :]
adata = adata[adata.obs.pct_counts_mt < 5, :]
normalize_total 将数据矩阵 normalize 为每个细胞 10,000 个 reads,从而使 counts 在各个细胞之间具有可比性。
sc.pp.normalize_total(adata, target_sum=1e4)
对数据进行对数运算:
sc.pp.log1p(adata)
识别高变基因
sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5)
sc.pl.highly_variable_genes(adata)

将 AnData 对象的 .raw 属性设置为 normalized 和 logarithmized 的原始基因表达,以便稍后在基因表达的差异测试和可视化中使用。这只是冻结了 AnData 对象的状态。
您可以通过调用 .raw.to_data() 来获取 .raw 中对象的 AnnData。
adata.raw = adata
如果您不在下面使用 sc.pp.regression_out correcting 数据并通过 sc.pp.scale 进行 scaling,那么您也可以完全不使用 .raw。
先前的高变基因检测的结果作为注释存储在 .var.highly_variable 中,并由 PCA 自动检测,从而由 sc.pp.neighbors 和随后的流形/图工具自动检测。在这种情况下,实际上进行以下过滤的步骤也是不必要的。
实际进行过滤
adata = adata[:, adata.var.highly_variable]
对每个细胞的总计数和线粒体基因表达的百分比的影响进行回归。将数据缩放为单位方差。
sc.pp.regress_out(adata, ['total_counts', 'pct_counts_mt'])
将每个基因按单位方差进行缩放。剪裁值超过标准偏差 10。
sc.pp.scale(adata, max_value=10)
2 主成分分析
通过运行主成分分析(PCA)来降低数据的维度,主成分分析揭示了变化的主轴并对数据进行去噪。
sc.tl.pca(adata, svd_solver='arpack')
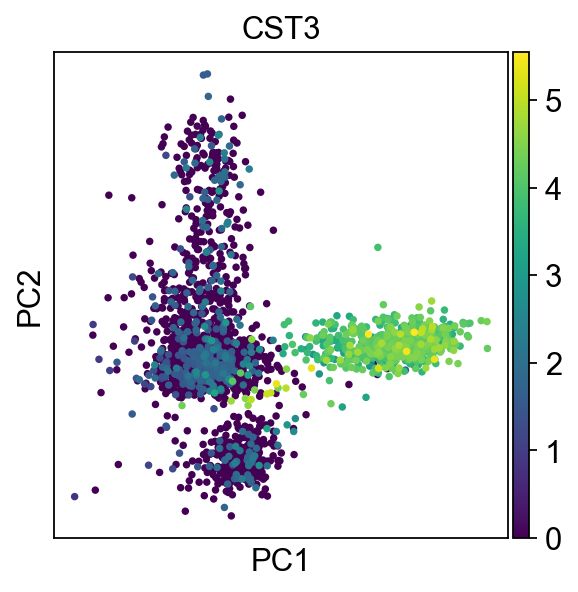
我们可以在 PCA 坐标中绘制散点图,但以后不会使用。
sc.pl.pca(adata, color='CST3')
让我们检查单个 PC 对数据总方差的贡献。这为我们提供了关于我们应该考虑多少 PC 来计算细胞的邻域关系的信息,例如在聚类函数 sc.tl.louvain() 或 tSNE sc.tl.tSNE() 中使用。根据我们的经验,通常粗略估计 PCs 的数量是可以的。
sc.pl.pca_variance_ratio(adata, log=True)

保存结果
adata.write(results_file)
adata
## AnnData object with n_obs × n_vars = 2638 × 1838
## obs: 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt'
## var: 'gene_ids', 'n_cells', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'mean', 'std'
## uns: 'log1p', 'hvg', 'pca'
## obsm: 'X_pca'
## varm: 'PCs'
3 计算邻域图
让我们使用数据矩阵的 PCA 表示来计算细胞的邻域图。您可以在这里简单地使用默认值。为了重现 Seurat 的结果,让我们取以下值。
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=40)
4 嵌入邻域图
我们建议使用 UMAP 将图嵌入二维 (McInnes et al., 2018) ,见下文。它可能比 tSNE 更忠实于流形的全局连通性,也就是说,它更好地保留了轨迹。在某些场合中,您可能仍然会观察到断开连接的 clusters 和类似的连接冲突。它们通常可以通过运行来补救:
sc.tl.paga(adata)
sc.pl.paga(adata, plot=False) # remove `plot=False` if you want to see the coarse-grained graph
sc.tl.umap(adata, init_pos='paga')
sc.tl.umap(adata)
sc.pl.umap(adata, color=['CST3', 'NKG7', 'PPBP'])
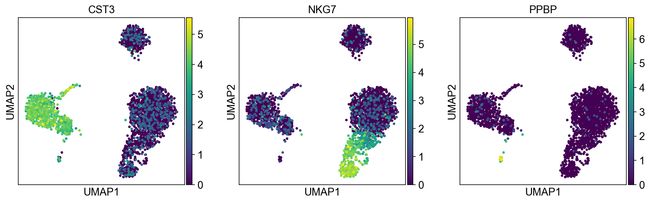
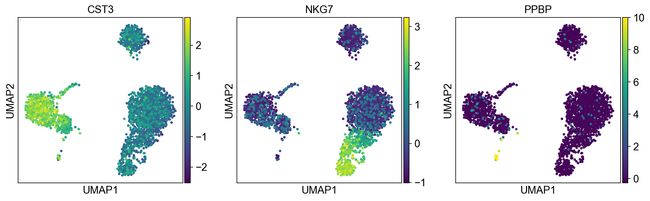
当我们设置 adata 的 .raw 属性时,前面的图显示了“raw”(normalized, logarithmized, but uncorrected)基因表达。你也可以通过明确声明你不想使用 .raw 来绘制 scaled 和 corrected 的基因表达。
sc.pl.umap(adata, color=['CST3', 'NKG7', 'PPBP'], use_raw=False)
5 对邻域图进行聚类
与 Seurat 和许多其他框架一样,我们推荐 Traag et al. (2018) 的 Leiden 图聚类方法(基于优化模块性的社区检测)。注意,Leiden 聚类直接对细胞的邻域图进行聚类,我们在上一节中已经计算过了。
sc.tl.leiden(adata)
绘制聚类图,这与 Seurat 的结果非常一致。
sc.pl.umap(adata, color=['leiden', 'CST3', 'NKG7'])

保存结果
adata.write(results_file)
6 寻找标记基因
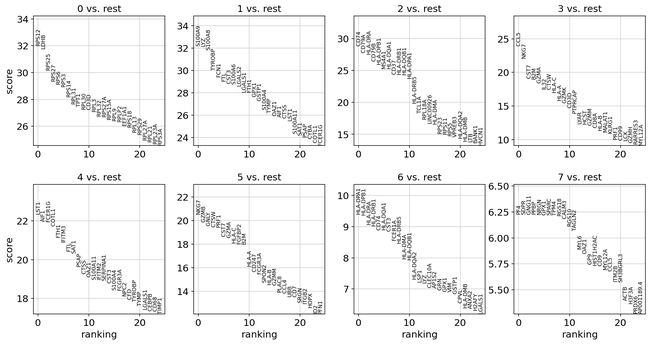
让我们计算每个聚类中高度差异基因的排名。为此,默认情况下,将使用 AnnData 的 .raw 属性,以防之前对其进行过初始化。最简单、最快的方法是 t-test。
sc.tl.rank_genes_groups(adata, 'leiden', method='t-test')
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)

sc.settings.verbosity = 2 # reduce the verbosity
Wilcoxon rank-sum (Mann-Whitney-U) 检验的结果非常相似。我们建议在出版物中使用后者,例如参见 Sonison & Robinson (2018)。您还可以考虑更强大的差异测试包,如 MAST、limma、DESeq2,对于python,还有最近的 diffxpy。
sc.tl.rank_genes_groups(adata, 'leiden', method='wilcoxon')
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)
保存结果
adata.write(results_file)
作为一种选择,让我们使用逻辑回归对基因进行排序。例如,Natranos et al. (2018) 提出了这一点。本质区别在于,在这里,我们使用多变量近似,而传统的微分测试是单变量的。Clark et al. (2014) 提供了更多细节。
sc.tl.rank_genes_groups(adata, 'leiden', method='logreg')
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)
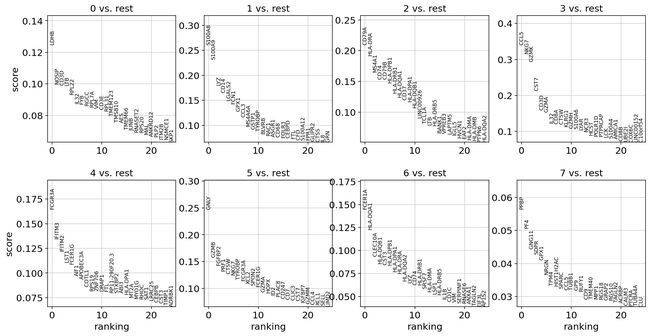
除了 IL7R(仅通过 t 检验发现)和 FCER1A(仅通过其他两种方法发现)之外,所有标记基因在所有方法中都被重复发现。
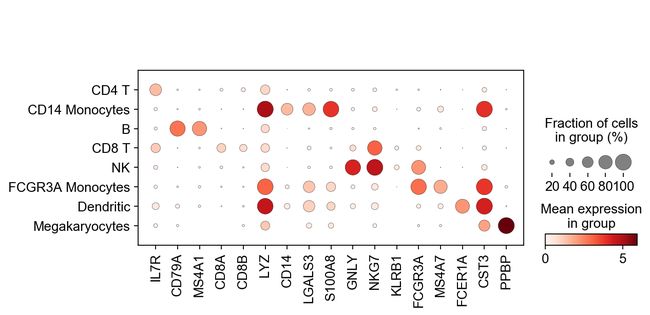
| Louvain Group | Markers | Cell Type |
|---|---|---|
| 0 | IL7R | CD4 T cells |
| 1 | CD14, LYZ | CD14+ Monocytes |
| 2 | MS4A1 | B cells |
| 3 | CD8A | CD8 T cells |
| 4 | GNLY, NKG7 | NK cells |
| 5 | FCGR3A, MS4A7 | FCGR3A+ Monocytes |
| 6 | FCER1A, CST3 | Dendritic Cells |
| 7 | PPBP | Megakaryocytes |
让我们也定义一个标记基因的列表,以供以后参考。
marker_genes = ['IL7R', 'CD79A', 'MS4A1', 'CD8A', 'CD8B', 'LYZ', 'CD14',
'LGALS3', 'S100A8', 'GNLY', 'NKG7', 'KLRB1',
'FCGR3A', 'MS4A7', 'FCER1A', 'CST3', 'PPBP']
使用 Wilcoxon Rank-Sum test 结果重新加载已保存的对象。
adata = sc.read(results_file)
在 dataframe 中显示每个 cluster 0、1、…、7 的 10 个排名靠前的基因。
pd.DataFrame(adata.uns['rank_genes_groups']['names']).head(5)
## 0 1 2 3 4 5 6 7
## 0 RPS12 S100A9 CD74 CCL5 LST1 NKG7 HLA-DPA1 PF4
## 1 LDHB LYZ CD79A NKG7 AIF1 GZMB HLA-DPB1 SDPR
## 2 RPS25 S100A8 HLA-DRA CST7 FCER1G GNLY HLA-DRA GNG11
## 3 RPS27 TYROBP CD79B B2M COTL1 CTSW HLA-DRB1 PPBP
## 4 RPS6 FCN1 HLA-DPB1 GZMA FTH1 PRF1 CD74 NRGN
找一张有分数和分组的表格。
result = adata.uns['rank_genes_groups']
groups = result['names'].dtype.names
pd.DataFrame(
{group + '_' + key[:1]: result[key][group]
for group in groups for key in ['names', 'pvals']}).head(5)
## 0_n 0_p 1_n 1_p 2_n 2_p 3_n 3_p 4_n 4_p 5_n 5_p 6_n 6_p 7_n 7_p
## 0 RPS12 1.238286e-224 S100A9 1.842655e-227 CD74 2.487145e-183 CCL5 6.969692e-127 LST1 1.419732e-110 NKG7 1.006113e-93 HLA-DPA1 5.422417e-21 PF4 4.722886e-10
## 1 LDHB 2.520407e-221 LYZ 8.413552e-227 CD79A 1.679730e-170 NKG7 3.765805e-108 AIF1 7.130846e-107 GZMB 5.598162e-88 HLA-DPB1 7.591860e-21 SDPR 4.733899e-10
## 2 RPS25 7.723960e-200 S100A8 1.257243e-222 HLA-DRA 6.942399e-167 CST7 1.187280e-81 FCER1G 1.045064e-106 GNLY 2.214727e-85 HLA-DRA 1.306768e-19 GNG11 4.733899e-10
## 3 RPS27 3.232559e-189 TYROBP 6.067012e-198 CD79B 2.569135e-154 B2M 2.397755e-80 COTL1 9.908055e-105 CTSW 2.404718e-84 HLA-DRB1 1.865104e-19 PPBP 4.744938e-10
## 4 RPS6 2.474817e-186 FCN1 4.894805e-190 HLA-DPB1 3.580735e-148 GZMA 5.599438e-76 FTH1 3.935567e-99 PRF1 5.451777e-84 CD74 5.853161e-19 NRGN 4.800511e-10
与单个 cluster 进行比较:
sc.tl.rank_genes_groups(adata, 'leiden', groups=['0'], reference='1', method='wilcoxon')
sc.pl.rank_genes_groups(adata, groups=['0'], n_genes=20)

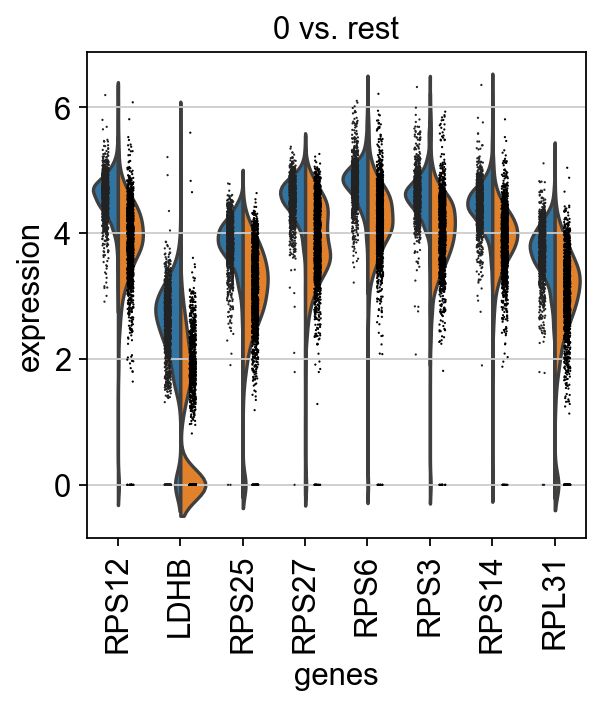
如果我们想要一个特定组的更详细的视图,请使用 sc.pl.rank_genes_groups_violin。
sc.pl.rank_genes_groups_violin(adata, groups='0', n_genes=8)

使用计算的微分表达式(即通过与其他组的比较来重新加载 DE)重新加载对象:
adata = sc.read(results_file)
sc.pl.rank_genes_groups_violin(adata, groups='0', n_genes=8)
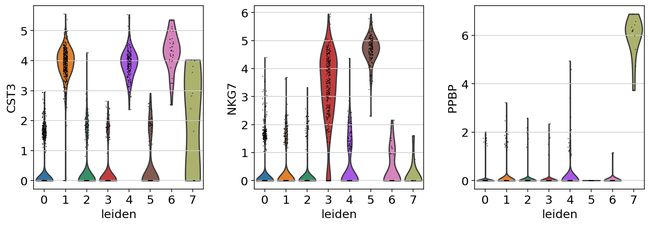
如果你想跨组比较某个基因,请使用以下方法。
sc.pl.violin(adata, ['CST3', 'NKG7', 'PPBP'], groupby='leiden')
实际标记细胞类型。
new_cluster_names = [
'CD4 T', 'CD14 Monocytes',
'B', 'CD8 T',
'NK', 'FCGR3A Monocytes',
'Dendritic', 'Megakaryocytes']
adata.rename_categories('leiden', new_cluster_names)
sc.pl.umap(adata, color='leiden', legend_loc='on data', title='', frameon=False, save='.pdf')

现在我们注释了细胞类型,让我们可视化标记基因。
sc.pl.dotplot(adata, marker_genes, groupby='leiden');
还有一个非常紧凑的小提琴图。
sc.pl.stacked_violin(adata, marker_genes, groupby='leiden', rotation=90);
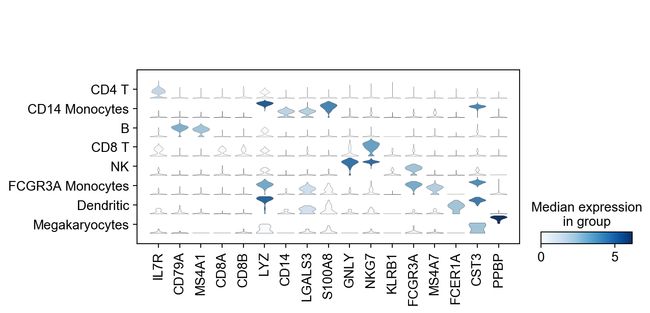
在分析过程中,AnData 积累了以下注释。
adata
## AnnData object with n_obs × n_vars = 2638 × 1838
## obs: 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt', 'leiden'
## var: 'gene_ids', 'n_cells', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'mean', 'std'
## uns: 'hvg', 'leiden', 'leiden_colors', 'neighbors', 'pca', 'rank_genes_groups', 'umap'
## obsm: 'X_pca', 'X_umap'
## varm: 'PCs'
## obsp: 'connectivities', 'distances'
adata.write(results_file, compression='gzip') # `compression='gzip'` saves disk space, but slows down writing and subsequent reading
使用 h5ls 大致了解该文件,它有许多选项-有关更多详细信息,请参阅此处。文件格式可能仍有待于未来的进一步优化。不过,所有读取功能都将保持向后兼容。
如果你想与那些只想将此文件用于可视化的人共享此文件,减少文件大小的一个简单方法是删除密集 scaled 和 corrected 的数据矩阵。该文件仍然包含 adata.raw 中可视化中使用的原始数据。
adata.raw.to_adata().write('./write/pbmc3k_withoutX.h5ad')
如果您想导出到“csv”,您有以下选项:
# Export single fields of the annotation of observations
# adata.obs[['n_counts', 'louvain_groups']].to_csv(
# './write/pbmc3k_corrected_louvain_groups.csv')
# Export single columns of the multidimensional annotation
# adata.obsm.to_df()[['X_pca1', 'X_pca2']].to_csv(
# './write/pbmc3k_corrected_X_pca.csv')
# Or export everything except the data using `.write_csvs`.
# Set `skip_data=False` if you also want to export the data.
# adata.write_csvs(results_file[:-5], )

本文由 mdnice 多平台发布