评估LLM在细胞数据上的实用性(3)-基因层面的评估
目录
- 定义
-
- 基因功能预测
- 扰动预测
- 基因网络分析
- 基因层面的评估
-
- 基因功能预测
- 扰动预测
- 基因网络分析
定义
基因功能预测
基因功能预测对于识别基因在不同条件下的特性非常重要。因为人类大约有20,000个蛋白质编码基因,只有一些被标注了功能。对基因功能的准确预测可以帮助我们理解和推断基因在生物系统中的作用。这里考虑基因的三种功能。第一种是剂量敏感或不敏感(dosage-sensitive or non-sensitive)。一些基因是剂量敏感的,这意味着它们在与遗传诊断相关的拷贝数变异(CNVs)分析中具有重要意义。第二个是二价和非甲基化(Bivalent versus non-methylated)。二价染色质结构是鉴定胚胎干细胞(ESCs)关键发育基因的重要手段。因此,鉴别二价标记基因和未甲基化基因是很重要的。第三个是二价和仅赖氨酸甲基化(Bivalent versus Lys4-only methylated)。Lys4-only methylated的基因也不同于二价标记的基因。我们借助真实的基因label比较模型的输出。我们把这个任务看作是一个二分类问题。这里使用了与细胞类型注释任务相同的metrics。scEval使用了一个公共数据集(Transfer learning enables predictions in network biology),并且只考虑数据集中的 labeled genes 进行预测和评估。
扰动预测
扰动预测(Perturbation Prediction)是一项基于基因编辑和单细胞测序技术的任务。在沉默一些基因后,可以通过测序获得未受干扰和受干扰的基因表达水平,这使我们可以探索基因之间的相互作用。一个著名的技术是Perturb-seq。在扰动预测中,我们计划预测基因编辑后的基因表达水平。该任务可以预测测试数据集中可见的基因扰动(比较简单),也可以预测测试数据集中不可见的基因扰动(比较困难)。
scEval把这个任务看作一个回归问题。在这里使用的度量是MPC(Mean Pearson Correlation),细胞 i i i的预测基因表达 g i ′ g_{i}' gi′和ground truth基因表达 g i g_{i} gi之间的Pearson correlation为: S P C i = g i ′ ⋅ g i ∣ ∣ g i ′ ∣ ∣ ⋅ ∣ ∣ g i ∣ ∣ S_{PC_{i}}=\frac{g'_{i}\cdot g_{i}}{||g_{i}'||\cdot||g_{i}||} SPCi=∣∣gi′∣∣⋅∣∣gi∣∣gi′⋅gi S M P C = ∑ i n S P C i n S_{MPC}=\frac{\sum_{i}^{n}S_{PC_{i}}}{n} SMPC=n∑inSPCi其中, S M P C S_{MPC} SMPC的值越大,说明表现越好。
在扰动预测任务中,通过选择非对照条件下的细胞(即受到某种基因指导操作影响的细胞),然后随机抽样对照条件下的细胞(即没有受到基因操作影响的正常细胞),将它们组合起来构建成一对input-target数据集,用作训练和测试数据集。其中,使用的Perturb-seq数据集来源于GEARS,包含了三种条件下的细胞数据:
- 对照(Control):没有进行基因扰动的细胞;
- 单基因扰动(One gene perturbation):进行了单个基因扰动的细胞;
- 双基因扰动(Two genes perturbation):同时进行了两个基因扰动的细胞。
在评估过程中,选择GEARS作为基准测试工具。
换句话说,为了训练和测试模型,研究者使用了GEARS数据集中的非对照细胞,这些细胞经历了一种或两种基因的扰动,然后与随机抽样的对照细胞结合。在模型评估阶段,单基因和双基因扰动的情况被视为一个整体,以检验模型在处理包含不同基因扰动情况的数据时的表现。这样做的目的是为了模拟和理解基因编辑技术如CRISPR对细胞状态的影响,并预测这些基因扰动可能导致的生物学变化。
基因网络分析
基因网络分析是下游任务。目的是推断特定的基因网络,例如,基因调控网络GRN或基因共表达网络GCN。GRN可以帮助理解基因之间的规律关系和预测的扰动结果。GCN可用于分析具有相似功能的基因或揭示某些疾病中基因的特征。GCN和GRN是两个不同的任务,因为相关性并不意味着因果关系。这一限制意味着我们不能仅仅根据嵌入的相似性或相关性来确定哪些基因是其他基因表达水平变化的“原因”。
在基因网络分析任务中,scEval使用真实基因和推断基因之间的重叠作为度量。比如,从特定的pathway中提取名称以HLA开头的基因和以CD开头的基因。此外,从基因embedding数据中提取具有显著相关性的基因。然后计算 G p a t h G_{path} Gpath和 G n e t G_{net} Gnet两个基因集合的Jaccard相似度: J a c c a r d = ∣ G p a t h ∩ G n e t ∣ ∣ G p a t h ∪ G n e t ∣ Jaccard=\frac{|G_{path}\cap G_{net}|}{|G_{path}\cup G_{net}|} Jaccard=∣Gpath∪Gnet∣∣Gpath∩Gnet∣
基因层面的评估
基因功能预测
scEval考虑了Geneformer、scGPT和vanilla NN来完成这个任务。平均而言,Geneformer和scGPT在该任务中表现良好,Vanilla NN和scLLM之间存在很大性能差距。图4b显示了不同超参数设置下的accuracy。学习率和 loss weight 越小,结果越准确。与scGPT相比,Geneformer对Epoch更为敏感。对于scGPT,由于预训练在该任务中的贡献大于微调,因此增加epoch不影响模型性能。
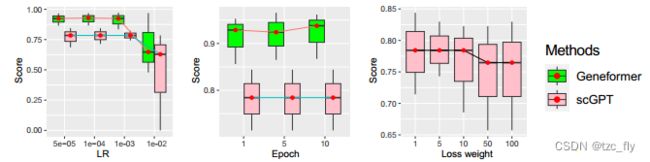
- 图4b:超参数对基因功能预测的影响。
扰动预测
考虑了scGPT和GEARS来完成这个任务。在训练过程中,对受到扰动的基因进行了mask,试图重建输入细胞中所有基因的表达水平,而不仅仅是被屏蔽的基因。使用MPC作为度量来评估scGPT在不同超参数或初始设置下的性能。数据集包括两种扰动条件:单基因扰动和双基因扰动。根据实验,scGPT可以预测具有较高MPC的基因扰动。
图4c总结了Norman、Adamson和Dixit数据集不同初始设置下的scGPT结果。在不同的设置中,默认设置对这些数据集的性能最好。这表明scGPT的初始配置可以很好地执行此任务。
关于超参数的影响,scGPT对学习率和epoch的调整非常敏感。降低学习率和增加epoch数可以改善MPC。其余的超参数在这个任务中贡献不大。此外,scEval发现任务特定损失分量是扰动预测的重要设计。
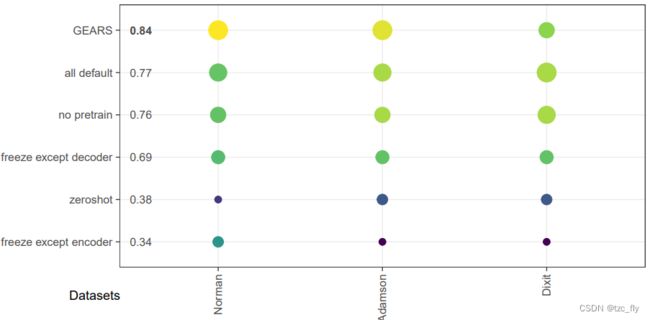
- 图4c:GEARs和scGPT(不同设置)在不同数据集上的表现。MPC越高意味着性能越好。
基因网络分析
在scGPT中的是GCN,因为构建过程是基于嵌入相似度的。在使用scGPT推断GCNs的过程中,定义了两种类型的GCN:
- Type 1 GCN,Tissue-specific GCN:这种类型的GCN是在零样本学习框架下,将scGPT模型应用于整个数据集,生成基因嵌入。然后计算Pearson相关性来推断基于这些嵌入的基因-基因关系。GCN的质量是根据不同细胞类型的标记基因之间的关系来评估的。
- Type 2 GCN,Cell-type specific GCN:该GCN是在零样本学习框架下,应用scGPT模型生成细胞类型特定的基因嵌入,并基于这些嵌入利用余弦相似度来推断基因-基因关系。该GCN的质量是基于针对细胞类型特定的基因集的基因本体富集分析(Gene Ontology Enrichment Analysis,GOEA)来评估的。
这些GCNs可以为理解特定组织或细胞类型中的基因相互作用和调控提供有价值的见解,这可能在生物学和医学中具有广泛的应用。
scEval使用免疫人类图谱数据集来评估推断这两种类型的GCNs的性能。由于我们知道该数据集的基因富集信息是先验知识,因此可以基于该数据集评估scGPT的性能。利用标记基因、细胞类型和GO通路等已知信息来评估scGPT推断的GCNs的性能。
在对免疫人类图谱数据集的分析中,scEval还考虑了另一种定义GCN的方法,类似于scGPT的定义。考虑根据基因嵌入来寻找基因的最近邻居。最初的重点是Type 1 GCN,结果如图4d和4e所示。根据标记基因的分布,该数据中的相邻关系被着色。根据免疫人类图谱数据集原论文收集标记基因。
图4d中,只有来自两种细胞类型的标记基因表现出共嵌入和分离关系。它们是单核细胞衍生的树突状细胞(Monocyte-derived dendritic cells)和巨核细胞祖细胞(Megakaryocyte progenitors)。另一方面,图4e表示基于Leiden的聚类标签。这些簇可以被解释为具有共同功能的基因群。对于其他细胞类型的标记基因,有的位于不同的簇中,如图4e所示,有的基因与其他细胞类型的标记基因共嵌入。有两个分离的簇(9和12),但在这两个簇中没有发现标记基因。
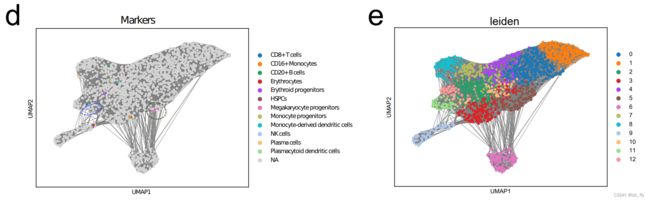
- 图4d:用不同细胞类型的标记基因着色基因embedding相似度网络。图4e:对基因embedding聚类。
注意,scEval中没有对GRN开展实验。