使用 ChatGPT 为生物信息学初学者赋能
论文:Empowering Beginners in Bioinformatics with ChatGPT. 2023
对于生信初学者而言,最大的困难是身边没有经验丰富的人给予指导。而ChatGTP的出现可能改变这一现状,学生可以自己作为导师,指导ChatGPT完成数据分析工作。
众所周知,与ChatGPT互动,给予的指令越精确,那么它给出的答案越精准。这篇论文提出一个与ChatGPT互动的模型:OPTICAL。其基本思想是通过迭代不断优化给予ChatGPT的指令。
该模型的流程图如下:
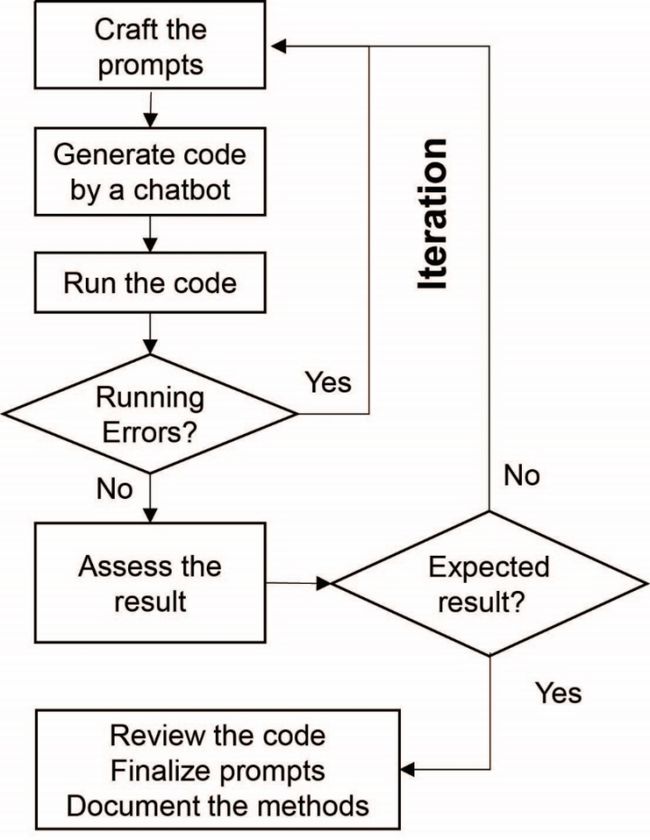
给予初始提示。
机器人产生分析代码。
运行代码。
如果出现错误,转向优化提示词。
如果代码正确,继续下一步。
评估结果。
如果结果不符合预期,转向优化提示词。
如果结果符合预期,继续下一步。
审查代码,得到最终提示词并归档方法。
这个模型本身平平无奇,符合平常人们使用ChatGPT的习惯:即不断优化提示词,直至得到正确答案。下面两个案例很好地体现了这一过程。
案例一:下一代测序的短读段比对和视觉检查
定义聊天机器人的行为:
Act as an experienced bioinformatician proficient in ChIP-Seq data analysis, you will assist me by writing code with number of lines as minimal as possible. Rest the thread if asked to. Reply “YES” if understand.
迭代0
I have two fastq files in current folder from single-end sequencing of a ChIP-Seq library: ENCFF000AVS_1m.fastq.gz, and ENCFF000AVS_10m.fastq.gz. For each fastq file, align reads to the human reference genome, save to bam file, and then covert it to bigwig file. Tools to use: bowtie2, samtools, and deepTools. The index for bowtie2 is in the folder “../data/indx/bowtie2_whole_genome/” with “hg38” as the prefix. Use 24 CPU for the alignment. Please draft the code in bash.
迭代1
[E::idx_find_and_load] Could not retrieve index file for 'ENCFF000AVS_1m.bam'
迭代2
Wait, I saw that you have "samtools index" before "bamcoverage". Does bamcoverage as bam to be sorted before using as input?
审查代码
I need to insert line-by-line comments to the below code which works well to address the needs for the data analysis task. Wait for my code.
最终提示词(粗体字是经过迭代加入的提示细节):
Act as an experienced bioinformatician proficient in ChIP-Seq data analysis, you will assist me by writing code with number of lines as minimal as possible. Rest the thread if asked to. Reply “YES” if understand.
I have two fastq files in current folder from single-end sequencing of a ChIP-Seq library: ENCFF000AVS_1m.fastq.gz, and ENCFF000AVS_10m.fastq.gz. For each fastq file, align reads to the human reference genome, save to bam file, index it, and then covert it to bigwig file with CPM normalization. Tools to use: bowtie2, samtools, and deepTools. The index for bowtie2 is in the folder “../data/indx/bowtie2_whole_genome/” with “hg38” as the prefix. Use 24 CPU for the alignment. Please draft the code in bash.
安全二:推断DNA序列的分子进化系统发育树
定义聊天机器人的行为:
Act as an experienced bioinformatician proficient in R, you will write code with number of lines as minimal as possible. Rest the thread if asked to. Reply “YES” if understand.
迭代0
You have a multiple alignment file named as tp53.clustal in ClustalW format. Please write R code that can load the file, calculate evolutionary distance, build a NJ tree, and visualize the phylogeny.
迭代1
I got an error message complaining "could not find function "read.alignment". Please fix it.
迭代2
I got a warning message " In dist.dna(aln) : NAs introduced by coercion". Please fix it.
迭代3
I wrote an R program to read a multiple alignment file named as tp53.clustal in ClustalW format, calculate evolutionary distance, build a NJ tree, and visualize the phylogeny. But I want to root the tree with the Zebrafish sequence as the outgroup. Can you help me revise the R code? Below is my R code.
# Load the required packages
library(seqinr)
library(ape)
# Read in the alignment file
aln <- read.alignment("tp53.clustal", format="clustal")
# Calculate the evolutionary distance
dist <- dist.dna(as.DNAbin(aln))
# Build the NJ tree
tree <- nj(dist)
# Plot the phylogeny
plot(tree)迭代4
I got an error message complaining " Error in nj(dist, outgroup = zebrafish_idx) unused argument (outgroup = zebrafish_idx)". Please fix it.
迭代5
I got an error message complaining "Error in if (newroot == ROOT) { : argument is of length zero". Please fix it.
审查代码
I created the following R code. Please add inline comments.
最终提示词
无。
关于简说基因
生信平台
Galaxy中国(UseGalaxy.cn)致力于打造中国人的云上生物信息基础设施。大量在线工具免费使用。无需安装,用完即走。活跃的用户社区,随时交流使用心得。
生信培训
简说基因的生信培训班,荣获学员的一致好评。如果你也对生物信息学感兴趣,欢迎来跟简说基因,学真生信。
生信分析
我们能够承接所有 NGS 组学数据分析业务,包括但不限于 WGS / WES / RNA-seq 等。基因组组装、注释,以及各种重测序业务都可以与简说基因合作。