QIIME2进阶五_QIIME2扩增子基因序列多样性分析
本节主要介绍了如何使用生物信息分析分析软件QIIME2对扩增子基因序列进行Alpha和Beta多样性分析,以及Alpha稀疏和深度选择。
本教程将使用来自人源化(humanized)小鼠的一组粪便样品,展示16S rRNA基因扩增子数据的“典型”QIIME 2分析。本教程旨在探讨人源化小鼠的遗传背景影响微生物群落的假设。
01 Alpha和Beta多样性分析Alpha and beta diversity analysis
--α多样性
香农(Shannon’s)多样性指数:群落丰富度的定量度量,即包括丰富度richness和均匀度evenness两个层面;
可观测的OTU:Observed OTUs,群落丰富度的定性度量,只包括丰富度;
Faith’s系统发育多样性:包含特征之间的系统发育关系的群落丰富度的定性度量;
均匀度Evenness:或 Pielou’s均匀度;群落均匀度的度量。
--β多样性
Jaccard距离:群落差异的定性度量,即只考虑种类,不考虑丰度;
Bray-Curtis距离:群落差异的定量度量,较常用;
非加权UniFrac距离:包含特征之间的系统发育关系的群落差异定性度量;
加权UniFrac距离:包含特征之间的系统发育关系的群落差异定量度量。
计算核心多样性命令:
time qiime diversity core-metrics-phylogenetic
--i-phylogeny rooted-tree.qza
--i-table table.qza
--p-sampling-depth 2000
--m-metadata-file sample-metadata.tsv
--output-dir core-metrics-results
| 命令注释 --p-sampling-depth:它是指定重采样(即稀疏/稀释)深度。这个脚本将随机地将每个样本的测序量重新采样至该参数提供的值。它将对每个样本中的计数进行无放回抽样,从而使得结果表中的每个样本的总计数为指定值。如果任何样本的总计数小于该值,那么这些样本将从多样性分析中删除。通过查看上面创建的表table.qzv文件中呈现的信息并选择一个尽可能高的值(因此每个样本保留更多的序列)同时尽可能少地排除样本来进行选择;--output-dir:将文件都输出到该目录下。 |
输出结果文件(13个数据文件):
core-metrics-results/faith_pd_vector.qza: Alpha多样性考虑进化的faith指数;
core-metrics-results/unweighted_unifrac_distance_matrix.qza: 无权重unifrac距离矩阵;
core-metrics-results/bray_curtis_pcoa_results.qza: 基于Bray-Curtis距离PCoA的结果;
core-metrics-results/shannon_vector.qza: Alpha多样性香农指数;
core-metrics-results/rarefied_table.qza: 等量重采样后的特征表;
core-metrics-results/weighted_unifrac_distance_matrix.qza: 有权重的unifrac距离矩阵;
core-metrics-results/jaccard_pcoa_results.qza: jaccard距离PCoA结果;
core-metrics-results/observed_otus_vector.qza: Alpha多样性observed otus指数;
core-metrics-results/weighted_unifrac_pcoa_results.qza: 基于有权重的unifrac距离PCoA结果;
core-metrics-results/jaccard_distance_matrix.qza: jaccard距离矩阵;
core-metrics-results/evenness_vector.qza: Alpha多样性均匀度指数;
core-metrics-results/bray_curtis_distance_matrix.qza: Bray-Curtis距离矩阵;
core-metrics-results/unweighted_unifrac_pcoa_results.qza: 无权重的unifrac距离的PCoA结果。
输出对象(4种可视化结果):
core-metrics-results/unweighted_unifrac_emperor.qzv: 无权重的unifrac距离PCoA结果采用emperor可视化;
core-metrics-results/jaccard_emperor.qzv: jaccard距离PCoA结果采用emperor可视化;
core-metrics-results/bray_curtis_emperor.qzv: Bray-Curtis距离PCoA结果采用emperor可视化;
core-metrics-results/weighted_unifrac_emperor.qzv: 有权重的unifrac距离PCoA结果采用emperor可视化。
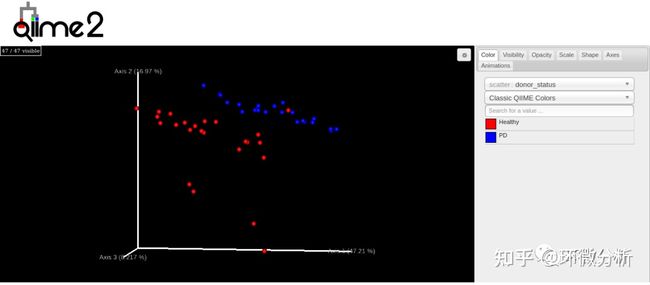
上图为有权重的unifrac距离PCoA结果采用emperor可视化结果。在可视化文件中可用鼠标拖动空间查看每个样本的分布位置。Axis1轴标签PCo 1 (47.21%)代表能最大区分所有样品的第一主坐标轴,可以解释样品中所有差异的47.21%;Axis2轴标签PCo 2 (16.97%)代表能最大区分所有样品的第二主坐标轴,可以解释样品中所有差异的16.97%;仅这两轴形成的第一个平面,即展示了样品间一半多的差异;Axis3轴标签PCo 3代表能最大区分所有样品的第三主坐标轴。每一个点代表一个样本,相同颜色的点来自同一个分组。两点之间距离越近表明两者的群落构成差异越小。将捐献者身份作为分类的依据,其中红色代表Healthy,蓝色代表PD。
| 命令注释当没有生成系统发育树时仍可进行核心多样性的计算,运行代码如下: qiime diversity core-metrics--i-table table.qza--p-sampling-depth xxx--m-metadata-file metadata.tsv --output-dir outdir |
Alpha多样性组间显著性分析和可视化命令:
time qiime diversity alpha-group-significance
--i-alpha-diversity core-metrics-results/faith_pd_vector.qza
--m-metadata-file metadata.tsv
--o-visualization core-metrics-results/faiths_pd_statistics.qzv
输出结果文件:core-metrics-results/faiths_pd_statistics.qzv,即faiths_pd指数按元数据的统计可视化。

上图主要分为三部分。上部箱线图展示以mouse_id为分类依据的alpha多样性,纵轴为faiths_pd指数,分类依据可在column选项卡中选择;中部为所有组别之间的KW检验结果,p值约为0.067,说明差异不显著;下部为两组之间的KW检验结果,435和437两组之间的p值约为0.149,说明两组之间差异不显著,其余各组之间分析同理。
用方差分析(ANOVA)来测试多重效应是否显着影响α多样性,命令如下:
time qiime longitudinal anova
--m-metadata-file core-metrics-results/faith_pd_vector.qza
--m-metadata-file metadata.tsv
--p-formula 'faith_pd ~ genotype * donor_status'
--o-visualization core-metrics-results/faiths_pd_anova.qzv
输出结果文件:core-metrics-results/faiths_pd_anova.qzv,即faiths_pd指数按元数据分组交互计算的anova统计可视化。

上图分为三部分。上部为模型结果,数值分别为平方和、自由度、两个均方的比值F和P值,F越大说明差异越明显;中部为预计残差图,展示了真实值和预测值之间的差异;下部为成对t检验的结果。
Beta多样性组间显著性分析和可视化,命令如下:
qiime diversity beta-group-significance
--i-distance-matrix core-metrics-results/unweighted_unifrac_distance_matrix.qza
--m-metadata-file metadata.tsv
--m-metadata-column cage_id
--o-visualization core-metrics-results/unweighted-unifrac-cage-significance.qzv
--p-pairwise
| 参数注释--p-pairwise:是基于置换检验的,用来检查差异组之间是否存在个体差异。--m-metadata-column:元数据文件中被选中用作分析的列。 |
置换检验:是指随机化检验或重随机化检验。对原始数据分布没有要求,特别适用于不满足于传统分析方法的条件,如小样本数据。
输出结果文件:core-metrics-results/weighted-unifrac-cage-significance.qzv,即按笼子统计有权重unifrac距离的显著性。

检验采用的是PERMANOVA方法。PERMANOVA分析(也叫NPMANOVA、Adonis分析)是一种以距离矩阵为对象的多元方差分析,非参数多元方差分析。检验组间群落差异本质上是检验距离矩阵之间的差异,普通的ANOVA分析无能为力。而基于距离矩阵的PERMANOVA分析则可以。在上图中显示p=0.001,说明差异极显著。同时也说明受捐赠者的影响显著。在下方的表格中还可查看两个笼子之间的p值。
检查笼子相关的差异是否是由于笼内较大差异引起,命令如下:
time qiime diversity beta-group-significance
--i-distance-matrix core-metrics-results/weighted_unifrac_distance_matrix.qza
--m-metadata-file metadata.tsv
--m-metadata-column cage_id
--o-visualization core-metrics-results/unweighted-unifrac-cage-significance_disp.qzv
--p-method permdisp
输出结果文件:core-metrics-results/unweighted-unifrac-cage-significance_disp.qzv,即按笼子统计无权重unifrac距离的显著性。

使用--p-method标志来使用permdisp,可基于cage_id的离散度,以检查笼子相关的差异是否是由于笼内较大差异引起。
多变量数据统计分析(PERMDISP):是一种Beta多样性支持的统计检验方法,除此之外还有ANOSIM和PERMANOVA。
查看供体和基因型之间的交集,命令如下:
time qiime diversity adonis
--i-distance-matrix core-metrics-results/unweighted_unifrac_distance_matrix.qza
--m-metadata-file metadata.tsv
--o-visualization core-metrics-results/unweighted_adonis.qzv
--p-formula genotype+donor
输出结果文件:core-metrics-results/unweighted_adonis.qzv,即供体和基因型交互条件统计无权重unifrac距离的显著性。
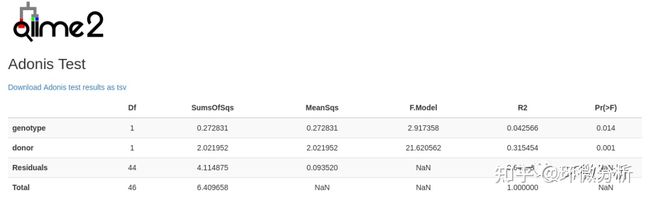
上图为常规的方差分析结果,可以用于比较组间变量的差异水平。Pr(>F)的值小于0.05时说明存在差异,值越小差异越大。
02 Alpha稀疏和深度选择 Alpha Rarefaction and Selecting a Rarefaction Depth
微生物多样性分析中需要验证测序数据量是否足以反映样品中的物种多样性,稀释曲线(丰富度曲线)可以用来检验这一指标,并间接反映物种的丰富程度。具体方法为:利用已测得16S rDNA序列中已知的各种OTU的相对比例,;来计算抽取n个(n小于测得reads序列总数)reads时出现OTU数值的期望值,然后根据一组n值(一般为一组小于总序列数的等差数列)与其相对应的OTU数量的期望值做出曲线。当曲线趋于平缓或达到平台期时也就可以认为测序深度已经基本覆盖到样品中所有的物种;反之,则表示样品中物种多样性较高,还存在较多未被测序的物种。运行命令如下:
time qiime diversity alpha-rarefaction
--i-table table.qza
--i-phylogeny tree.qza
--p-max-depth 4250
--m-metadata-file metadata.tsv
--o-visualization alpha-rarefaction.qzv
| 参数解读 --p-max-depth :重采样深度。提供的--p-max-depth参数的值应该通过查看上面创建的table.qzv文件中呈现的“每个样本的测序量”信息来确定。一般来说,选择一个在中位数附近的值似乎很好用。如果得到的稀疏图中的线看起来没有变平,那么应增加该值。如果由于大于最大采样深度而丢失了许多样本,则减少该值。--i-phylogeny:通常使用的是有根树。当使用fragment-insertion插件进行系统发育树构建时,Alpha稀疏曲线的构建则可省略此参数设置。--p-min-depth和--p-max-depth:在设置重采样深度时,可设置这两个参数来对特征表进行子采样。 |
输出结果文件:alpha_rarefaction_curves.qzv,即alpha稀疏曲线。
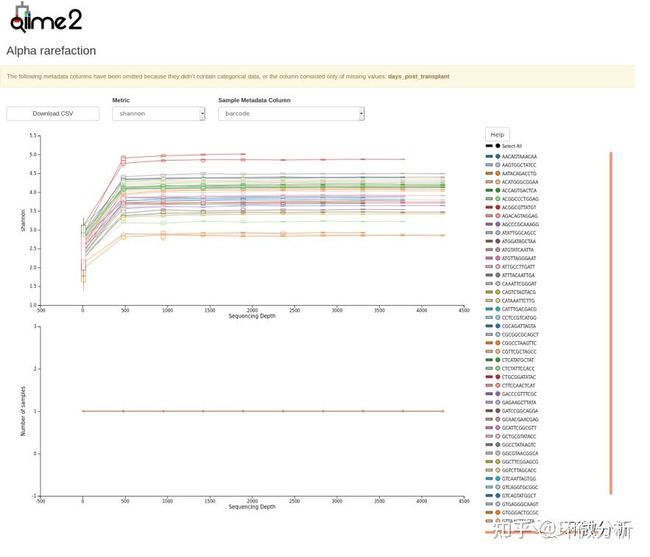
上图显示作为采样深度函数的α多样性(观察到的OTU或shannon),这用于判断丰富度或均匀度是否已饱和。当接近最大采样深度时,稀疏曲线应趋于平稳。反之可能表明样本中的丰富度尚未完全饱和。下图显示了每个采样深度的每个元数据类别组中的样本数。这对于确定样本丢失的采样深度以及元数据列组值是否存在偏差非常有用。当依据元数据进行分类时,下图尤为重要。
本文提供所有输出结果文件,百度网盘下载链接:
https://pan.baidu.com/s/1CSZgEaC66gIXjnHr_1GvdA
提取码:1234
这篇推文对你有帮助吗?喜欢这篇文章吗?喜欢就不要错过呀,关注本知乎号查看更多的环境微生物生信分析相关文章。亦可以用微信扫描下方二维码关注“环微分析”微信公众号,小编在里面载入了更加完善的学习资料供广大生信分析研究者爱好者参考学习,也希望读者们发现错误后予以指出,小编愿与诸君共同进步!!!

学习环境微生物分析,关注“环微分析”公众号,持续更新,开源免费,敬请关注!
转载自原创文章:
QIIME2进阶五_QIIME2扩增子基因序列多样性分析
最后,再次感谢你阅读本篇文章,真心希望对你有所帮助。感谢!