【中科院】分子生物学-朱玉贤第四版-笔记-第10讲 分子生物学操作技术
第 10 讲 分子生物学操作技术
文章目录
- 7. 分子生物学操作技术
-
- 7.1 DNA 基本操作技术
-
- 7.1.1 核酸的凝胶电泳
- 7.1.2 细菌转化 (transformation)
- 7.1.3 聚合酶链式反应 (PCR) 技术
- 7.1.4 实时定量 PCR (real time quantitative PCR,Q-PCR)
- 7.1.5 重亚硫酸盐测序技术 (Bisulfite Sequencing)
- 7.1.6 基因组 DNA 文库构建
- 7.2 RNA 基本操作技术
-
- 7.2.1 总 RNA 的提取
- 7.2.2 mRNA 的纯化
- 7.2.3 cDNA 的合成
- 7.2.4 cDNA 文库的构建
- 7.2.5 基因文库的筛选
- 7.3 新基因的发现与克隆
-
- 7.3.1 简并引物
- 7.3.2 RACE(cDNA 末端快速扩增法)
- 7.3.3 RNA 的差异显示技术(DDRT-PCR) (此方法现已不用)
- 7.3.4 cDNA 差示分析法 (RDA)
- 7.3.5 Gateway 大规模克隆技术
- 7.3.6 基因的图位克隆法 (map-based cloning)
- 7.3.7 反向 PCR 技术
- 7.3.8 TAIL-PCR 克隆 T-DNA 插入位点侧翼序列
7. 分子生物学操作技术
如何学习分生操作技术
- 技术的原理
- 应用范围
- 优缺点
核酸基本操作技术的应用需求
- 分离
- 大小鉴定
- 序列鉴定
- 扩增
- 标记
7.1 DNA 基本操作技术
7.1.1 核酸的凝胶电泳
● 琼脂糖 (agarose) 和聚丙烯酰胺 (polyacrylamide) 凝胶电泳技术,能按分子量大小分离 DNA, 是分析鉴定重组 DNA 分子及蛋白质与核酸相互作用的重要实验手段——分离和确定大小。
凝胶电泳技术分离 DNA 片段的基本原理
- 一种分子被放置到电场中,它就会以一定的速度移向适当的电极。我们把这种电泳分子在电场作用下的迁移速度,叫做电泳的迁移率,它与电场强度和电泳分子本身所携带的净电荷数成正比,与片段大小成反比。
- 生理条件下,核酸分子中的磷酸基团呈离子化状态,所以,DNA 和 RNA 又被称为多聚阴离子 (polyanions) ,在电场中向正电极的方向迁移。
- 由于糖磷酸骨架在结构上的重复性质,相同数量的双链 DNA 几乎具有等量的净电荷,因此它们] 能以同样的速度向正电极方向迁移。
● 琼脂糖是琼脂的主要成分之一,是一种多糖,通常从某些红色海藻中提取。它是由琼脂二糖的重复单元组成的线性聚合物,琼脂二糖是由 D-半乳糖和 3,6-脱水-L-吡喃半乳糖组成的二糖。
● 聚丙烯酰胺是由丙烯酰胺亚单元形成的聚合物。它可以合成为简单的直链结构或交联,常用 N, N’-亚甲基双丙烯酰胺作为交联剂。具有高吸水性,在水合时形成软凝胶,用于聚丙烯酰胺凝胶电泳等应用。
琼脂糖及聚丙烯酰胺凝胶分辨 DNA 片段的能力

- 琼脂糖凝胶分辨 DNA 片段的范围为 0.2~50kb 之间。
- 聚丙烯酰胺凝胶的分辨范围为 1 到 1000 个碱基对之间。
- 凝胶浓度的高低影响凝胶介质孔隙的大小,浓度越高,孔隙越小,其分辨能力就越强。
电泳后 DNA 分子的显色 (结合 DNA 的染料)
- 溴化乙锭 (EB) 染料的化学结构及其对 DNA 分子的插入作用。由于插入了溴化乙锭分子,在紫外光照射下,琼脂糖凝胶电泳中 DNA 的条带便呈现出橘黄色荧光,易于鉴定。
- EB 的替代物:
①GoldView其实就是 AO (acridine orange)。 SIGMA 都有售;灵敏度差,对于 500bp 以下的片段效果不是很好,荧光基团在紫外灯下非常容易淬灭,一般 5-10min 条带就会消失。本底色严重,不利于回收,毒性很大,尤其在紫外灯下其诱导突变能力极高。
②Sybrgreen: 稳定性很差,毒性也很大,尤其在紫外灯下。有少许本底色。
③GelRed/GelGreen:稳定、灵敏度高,几乎没有底色,毒性低,价格贵 (1400rmb/0.5ml)
7.1.2 细菌转化 (transformation)
- 将外源 DNA 片段或者载体分子导入到寄主细菌细胞,菌株由于捕获了来自供体菌株或者外源 DNA 而导致性状特征发生遗传改变的过程。
- 外源 DNA 能在宿主细胞中通过自身载体上的复制起始位点进行复制增殖,从而在宿主细胞中长期保存,并以完整的形式从细胞中被分离纯化出来。
感受态细胞 (competent cells) 的制备
- 为提高效率,可对受体菌进行物理或化学处理,增加其获取 DNA 的能力。经过这种处理的细胞被称作感受态细胞 (
competent cells)。 - 转化方法:氯化钙法、电击法、基因工程噬菌体颗粒导入
★ CaCl2法(氯化钙法)
- 将快速生长期大肠杆菌置于经 0°C 预处理的低渗 CaCl2 溶液中,细胞膨胀,膜通透性改变,易与外源 DNA 相粘附。
- 将该体系转移到 42°C 下做短暂的热刺激,外源 DNA 就可能被细胞吸收。将经过转化后的细胞置于选择性培养基上,筛选阳性克隆。
- 转化效率可达到 5×106~2×107 个转化子/ug 超螺旋质粒 DNA。

● 电击法
- 电脉冲可以在细胞膜上造成小凹陷,形成疏水孔洞。随着跨膜电压增加,大疏水性孔洞会转变为亲水性孔洞,介质中的 DNA 进入细胞质。
- 将生长至对数中期的 E.coli 菌液冷却至 4°C 后离心,洗菌后用 10% 的甘油悬浮,将高密度菌液 (~2×1010/ml) 置于特制的电极杯中进行电击。
- 获得最大转化效率时场强一般为 12.5~15kV/cm, 时间跨度一般为 4.5~5.5 毫秒。
- 电击转化与温度有关,一般在 0~4°C 进行。由于转化载体上常带有 LacZ 基因,多用带有不同抗生素的选择性培养基结合 α-互补蓝白斑筛选法鉴定转化细胞。
● 基因工程噬菌体颗粒导入
- 利用噬菌体颗粒能够有效地将 DNA 注入到寄主细胞中这一特点,科学家还发明了重组体 DNA 分子的体外包装法。经包装的基因工程噬菌体颗粒能够借助细菌表面的噬菌体接受器位点将 DNA 注入受体细菌,并在这些细胞中得到繁殖和表达。
● 感受态细胞转化频率的计算方法
- 转化体总数 = 菌落数× (转化反应原液总体积 / 涂板菌液体积)
- 插人频率 = 白色菌落数 / (白色菌落数 + 蓝色菌落数)
- 转化频率(每微克 DNA 转化菌落数)= 转化体总数 / 加入质粒 DNA 总量 (ug)。
7.1.3 聚合酶链式反应 (PCR) 技术
- 聚合酶链式反应是快速扩增 DNA 序列最常用的方法。
- PCR 反应的模板 DNA 若是基因组上的某个片段,就称为
genomic PCR, 若是 mRNA 反转录产生的 cDNA, 就称为RT-PCR。 - 1983 年 8 月,凯利穆利斯 (Kary Mullis) 第一次在 公司正式作了一个有关 PCR 原理的学术报告。
1993 年因发明 PCR 技术,与迈克尔史密斯分享诺贝尔化学奖。
PCR 技术的原理
-
首先将双链 DNA 分子在临近沸点的温度下加热分离成两条单链 DNA 分子,DNA 聚合酶以单链 DNA 为模板并利用反应混合物中的四种脱氧核苷三磷酸 (dNTP)、合适的 Mg2+ 浓度和实验中提供的引物序列合成新生的 DNA 分子。
-
经不断重复循环之后,反应混合物中所含有的双链 DNA 分子数,即两条引物结合位点之间的 DNA 区段的拷贝数,理论上的最高值应是 2n,实得 1.8n。
-
PCR 步骤(一): DNA 解链(变性)
将含有待扩增 DNA 样品的反应混合物放置在高温 (>94°C) 环境下加热 1 分钟,使双链 DNA 变性,形成单链模板 DNA。

-
PCR 步骤(二): 引物与模板 DNA 相结合(退火)
降低反应温度(退火,约 50°C),约 1 分钟,使寡核苷酸引物与两条单链模板 DNA 结合在靶 DNA 区段两端的互补序列位置上。
控制引物浓度远大于模板量,防止双链 DNA 模板自身结合。
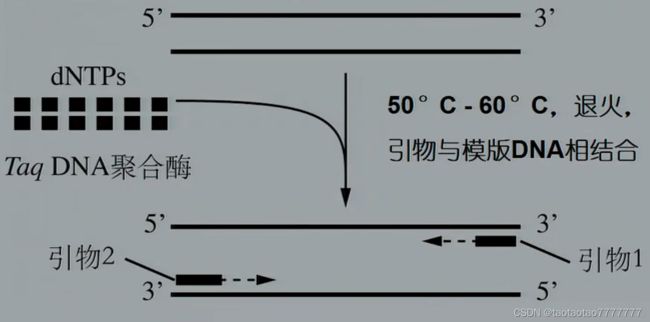
-
PCR 步骤(三) : DNA 合成(链的延伸)
将反应混合物的温度上升到 72°C 左右保温 1-数分钟,在 DNA 聚合酶的作用下,从引物的 3’端加入脱氧核苷三磷酸 dNTP,并沿着模板分子按 5→3"方向延伸,合成新生 DNA 互补链。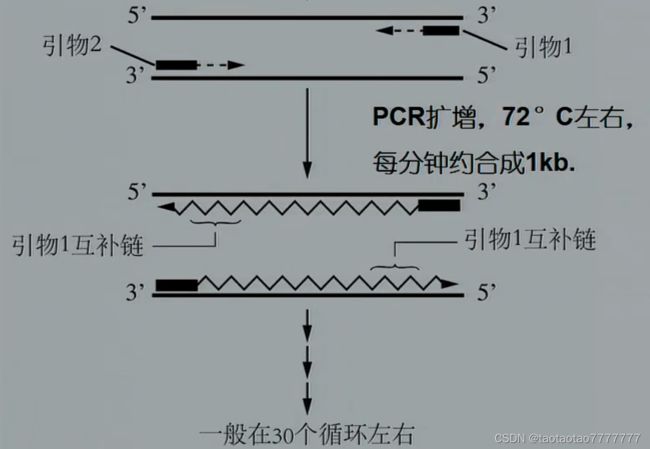
-
参考资料(b 站):引物设计与基因克隆
7.1.4 实时定量 PCR (real time quantitative PCR,Q-PCR)
由于 PCR 敏感性高,扩增产物总量变异系数大,定量不准确。90 年代末期出现了 Q-PCR, 利用荧光检测 PCR 仪绘制 DNA 扩增过程中的累积速率动态变化图,基本消除在测定终端产物丰度时变异系数较大的问题。
SYBR Green Ⅰ 探针
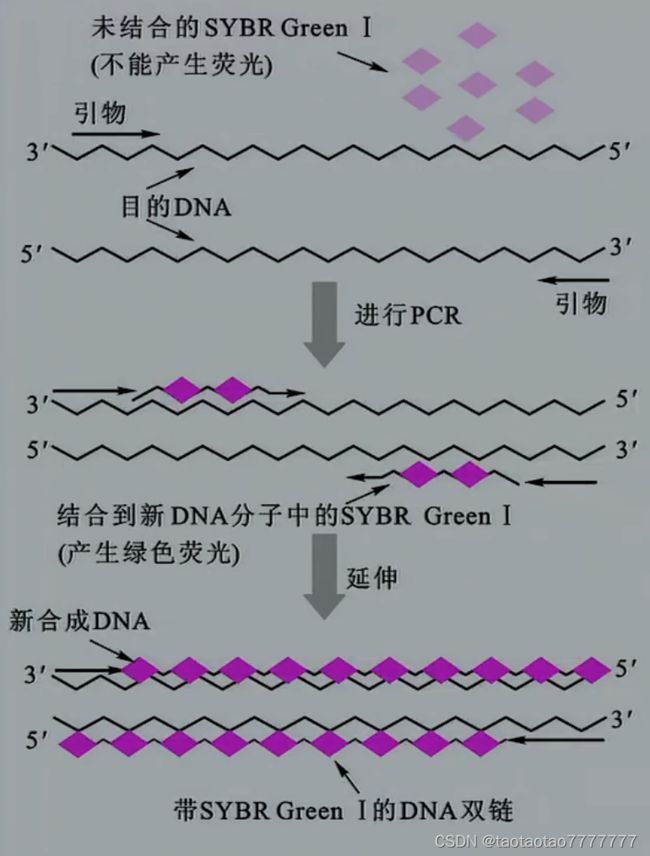
- 非序列特异性荧光染料
SYBR Green Ⅰ,激发光波长 520nm,这种荧光染料 只能与双链 DNA 结合。 - 混合在 PCR 反应液中的荧光探针只有与大片段 DNA 结合后,才能够被激发出荧光。
- 随着新合成 DNA 片段的增加,由于结合到 DNA 上的荧光探针增加,被激发产生的荧光相应增加。
用实时定量 PCR 法分析未知样品靶基因的绝对量

Ct 值是产物荧光强度首次超过设定阈值时,PCR 反应所需的循环数。利用标准曲线,可以确定样品中待检测靶 DNA 的绝对含量。- 实时定量 PCR 相对定量实验的数据处理
下图拟南芥野生型和突变体样品经由拟南芥持家基因 (UBQ10) 进行均一化处理,突变型 WUS 基因的表达丰度比野生型提高了 239 倍。


TaqMan 探针
- 利用 SYBR Green Ⅰ可以检测 PCR 反应中获得的全部双链 DNA,但是不能区分不同的双链 DNA。为了进一步确保荧光检测的就是靶 DNA 序列,人们又设计了仅能与目的 DNA 序列特异结合的荧光探针,如 TaqMan 探针。
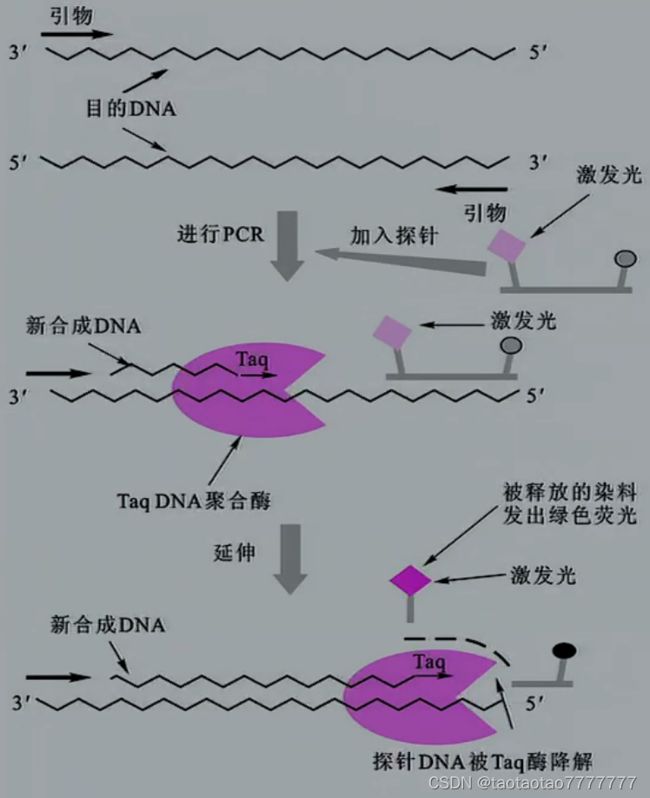
TaqMan 探针是一小段被设计成可以与靶 DNA 序列中间部位结合的单链 DNA(一般为 50~150 bp),并且该单链 DNA 的 5’和 3’端带有短波长(菱形)和长波长(小圆形)两个不同荧光基团。这两个荧光基团由于距离过近,在荧光共振能量转移 (FRET) 作用下发生了荧光淬灭,因而检测不到绿色荧光。- PCR 反应开始后,随着 PCR 反应的进行,TaqMan 探针结合到靶 DNA 序列上,当 TaqDNA 聚合酶延伸链时,它的核酸外切酶活性会逐个切掉结合在目的 DNA 中部的 TaqMan 探针的碱基,从而使两个荧光基团被释放出来,解除了荧光淬灭的束缚。短波长的黄光基团在激发光下产生绿色荧光,它的荧光强度反映新合成目的 DNA 的产量。
7.1.5 重亚硫酸盐测序技术 (Bisulfite Sequencing)
- DNA 分子上可能发生多种化学修饰,如甲基化、乙酰化等。在不改变 DNA 序列的情况下,通过对 DNA 分子的化学修饰,可以改变基因表达水平。相对于传统的遗传学——即只有 DNA 序列变化才能导致基因表达的改变——而言, 这种现象被称为表观遗传学 (
Epigenetics) 。
DNA 甲基化
- DNA 甲基化 (
DNA methylation) 是最早发现的基因的表观修饰方式之一,具体指生物体在 DNA 甲基转移酶 ( DNA methyltransferase,DNMT) 的催化下,以 S-腺苷甲硫氨酸 (SAM) 为甲基供体将甲基转移到特定的碱基上的过程。 - 甲基化的主要形式有 5-甲基胞嘧啶 (
m5C)、6-甲基腺嘌呤 (m6A) 和 7-甲基鸟嘌呤 (m7G),在 真核生物中甲基化主要发生于胞嘧啶 C。 - DNA 甲基化能引起染色质结构、DNA 构象、DNA 稳定性及 DNA 与蛋白质相互作用方式的改变,从而参与调控许多重要的生物学现象和发育过程。通常,DNA 的甲基化会抑制基因表达。
- 在动物细胞中,有三种 DNA 甲基化状态:① 持续的低甲基化状态,如管家基因;② 去甲基化状态,如一些发育阶段特异表达基因;③ 高度甲基化状态,如女性的一条失活的 X 染色体。
- DNA 的 CpG 序列在动物基因组中出现的频率仅有 1%,远低于基因组中的其他双核苷酸序列。但在基因组的某些区域中,CpG 序列密度可达均值的 5 倍以上,成为鸟嘌呤和胞嘧啶的富集区,形成所谓的 CpG 岛 (
CpG island)。哺乳动物基因组中约有 4 万个 CpG 岛。一般情况下,只有 CpG 岛中的胞嘧啶能够被甲基化,CpG 岛通常位于基因的 启动子区或第一个外显子区。 - 当肿瘤发生时,抑癌基因 CpG 岛以外的 CpG 序列非甲基化程度增加,而 CpG 岛中的 CpG 呈高度甲基化状态,使得染色体螺旋程度增加,抑癌基因不能表达。
- 植物细胞 DNA 甲基化的范围明显大于动物细胞。发生甲基化的位点不再限于 CpG 岛(在 CHG 和 CHH 位点都有发生,H=A、T 或 C), 发生甲基化的位置也不再局限于启动子区和第一外显子区。
常用确定某个碱基位点的甲基化情况的实验方法
- 甲基化敏感限制性内切核酸酶法 (melhylation sensitive rstriction endonuclease,
MS-RE) - 重亚硫酸盐测序法 (bisulfite sequencing)
- 甲基化特异性的 PCR (methylation-specific PCR,
MS-PCR) - DNA 微阵列法
- 甲基化敏感性斑点分析法 (methylation sensitive dot blot assay,
MS-DBA)
重亚硫酸盐测序 (bisulfite sequencing)

● 主要实验过程:
- 将待测 DNA 样品用限制性内切酶处理或超声波破碎等物理方法打断成 500~1000 bp 的碎片,
- 重亚硫酸盐处理使 DNA 中未甲基化的胞嘧啶 C 发生脱氨基变成尿嘧啶 U , PCR 扩增后被测序仪读为胸腺嘧啶 T。
- 已甲基化的胞嘧啶由于甲基的保护而不受影响。
- 参考原始序列判定原 C 位点是否甲基化——未甲基化的 C 位点变为 T, 甲基化的 C 位点仍保持为 C。
● 注意
- 与普通 PCR 不同,没有甲基保护的 C 在重亚硫酸盐处理后就会转变为 U, 设计重亚硫酸盐测序引物时就要将该位点相应改为 T。
- 由于 DNA 分子上的 C 位点通常不是 100% 的甲基化或非甲基化,所以合成引物时常常不可避免地要出现 简并位点,正向引物中为 Y(Y=C 或 T),反向引物中简并位点 R(R=G 或 A)。
- 重亚硫酸盐测序分析时,必须对同一目标片段进行多次测序,通常要求==至少测序 11 次==,以避免产生同源测序。所谓同源测序 (
sibling sequencing), 是指实验中所挑取的克隆来源于同一个原始 DNA 模板分子,形成了完全相同的没有代表性的甲基化模式。 - 要设置已知甲基化水平的正对照。
- 由于重亚硫酸盐测序的引物常有简并位点,造成 PCR 扩增过程中引物对不同甲基化程度 DNA 模板分子亲和力的不同,使亲和力高的模板分子在 PCR 终产物中占高比例,所以实验中经常用其他方法做平行验证。
● 不同陆地棉样品 KCS 13 启动子区重亚硫酸盐测序结果(17 克隆)
- 取不同陆地棉纤维基因组 DNA 进行重亚硫酸盐测序。每个样品测 17 个独立克隆(17 行)。每个小圆圈表示该段序列的一个 C 位点。
- 实心圆:甲基化状态
- 空心圆:非甲基化状态
- 箭头:甲基化水平发生明显变化(找到的 6 个甲基化水平发生变化的 CHH 位点)

- 该区段共包含 41 个 C 位点,包括 2 个 CG 位点(红色)、8 个 CHG 位点(蓝色)和 31 个 CHH 位点(绿色)。
- 在 A 和 B 两个不同的棉花纤维基因组 DNA 样品中,该启动子区段的 2 个 CG 位点和 7 个 CHG 位点保持了高甲基化状态 (>76%),1 个 CHG 位点在两个样品中都保持非甲基化状态;而在 CHH 位点上的甲基化水平发生了显著变化(A 甲基化→B 非甲基化)。
- 由于重亚硫酸盐测序的引物常有简并位点,造成 PCR 扩增过程中引物对不同甲基化程度 DNA 模板分子亲和力的不同,使亲和力高的模板分子在 PCR 终产物中占高比例,所以实验中经常用其他方法做平行验证。
7.1.6 基因组 DNA 文库构建
- 把某种生物的基因组 DNA 切成适当大小,分别与载体组合,导入微生物细胞,形成克隆。汇集包含基因组中所有 DNA 序列的克隆(理论上每个序列至少有一个代表),这样的克隆片段的总汇,称为
基因组 DNA 文库。 - 用途:分离特定的基因片段、分析特定基因结构、研究基因表达调控、构建全基因组物理图谱、全基因组序列测定。
- 构建基因组 DNA 文库的第一步是制备大小合适的随机 DNA 片段,在体外将这些 DNA 片段与适当的载体相连成重组子,转化到大肠杆菌或其他受体细胞中,从转化子克隆群中筛选出含有靶基因的克隆。
- 为保证能从基因组文库中筛选到某个特定基因,基因组文库必须具有一定的代表性和随机性。也就是说文库中全部克隆所携带的 DNA 片段必须覆盖整个基因组。
- 在文库构建中通常采用两种策略提高文库代表性:一是用机械切割法或限制性内切核酸酶切割法随机断裂 DNA, 以保证克隆的随机性;二是增加文库重组克隆的数目,以提高覆盖基因组的倍数。
- 预测一个完整基因组文库应包含的克隆数目,可用 Clark 和 Carbon 于 1975 年提出的公式:N = ln(1-p) / ln(1-f)
- N 表示一个基因组文库所应该包含的重组克隆数目
- p 表示所期望的靶基因在文库中出现的概率
- f 表示重组克隆平均插入长度与基因组 DNA 总长之比。
- 以人为例,其基因组大小为 3x109 bp, 若要求 p = 99%,平均插入片段大小为 20 kb, 则 N= 6.9×105。
利用λ噬菌体载体构建基因组文库
-
构建基因组文库最常用的是 λ噬菌体载体(克隆能力约 15—20kb)和限制性内切酶部分消化法。
-
例如用识别 4 个核苷酸的核酸限制性内切酶 Sau3A,与 BamHⅠ 是一对同尾酶消化 DNA,由 Sau3A 酶切产生的 DNA 片段可插入到经 BamHⅠ 消化的λ噬菌体载体上。

-
(基本流程如图所示)提取真核基因组 DNA,用 Sau3A 局部消化,消化产物经琼脂糖凝胶电泳或者蔗糖梯度离心,收集相对分子质量为 15 ~20 kb 范围的 DNA 片段。同时用 BamHⅠ 消化 λ 噬菌体载体 DNA,纯化后用 T4 连接酶与收集的 DNA 片段相连接,形成嵌合分子。体外包装后用重组噬菌体感染大肠杆菌受体细胞,产生噬菌斑,组成包含该真核生物基因组绝大部分序列的 DNA 文库。λ 噬菌体文库构建方法简单高效,所获得的文库易于用分子杂交法进行筛选,因此被广泛用于细菌、真菌等基因组较小物种的研究。
-
此外,还有很多大容量克隆载体,如柯斯质粒、细菌人工染色体 (BAC)、P1 源人工染色体 (PAC)、酵母人工染色体 (YAC) 等都可用于基因组文库的构建。
优点:可以插入更大片段 DNA;
缺点:稳定性一般较差,在大量复制的过程中容易出现缺失现象。操作也相对较困难。
7.2 RNA 基本操作技术
● 细胞中的总 RNA 包括 mRNA, rRNA ,tRNA 以及一些小 RNA (sRNA)。
● 一个典型的动物细胞约含 10-5ug RNA,其中:
- 80%-85% 为 rRNA
- 15%-20% 为 tRNA 及 sRNA
- mRNA 约占总 RNA 的 1%-5%
7.2.1 总 RNA 的提取
异硫氰酸肌–苯酚抽提法
- 总 RNA 的抽提方法有多种,目前实验室常用的方法是用异硫氰酸肌–苯酚抽提法。
- Trizol 试剂是使用最广泛的抽提 RNA 专用试剂,主要由苯酚和异硫氰酸胍组成,可以迅速破坏细胞结构,使存在于细胞质基质及核内的 RNA 释放出来,并使核糖体蛋白与 RNA 分子分离,还能保证 RNA 的完整。
- 提取 RNA 时,首先用液氮研磨材料,匀浆,加入 Trizol 试剂,进一步破碎细胞并溶解细胞成分。然后加入氯仿抽提,离心,分离水相和有机相,收集含有 RNA 的水相,通过异丙醇沉淀,获得比较纯的总 RNA, 用于下一步 mRNA 的纯化。
- 实验中还常将含有 RNA 样品的细胞破碎液通过一个硅胶膜纯化柱,使 RNA 吸附在硅胶膜上而与其他成分分开,进一步在低盐浓度下从硅胶膜上直接洗脱 RNA, 得到纯度较高的 RNA。
核酸浓度和纯度的测定

- 核酸在 240~290nm 的紫外波段有强烈的吸收峰
- 蛋白质(三个芳香氨基酸,有苯环共轭体系)也有吸收峰
- 浓度:OD260
- 纯度:OD260/OD280
★ DNA/RNA 浓度的定量测定
- No DNA/RNA: Optical Density=Log(100/100)=0
- 纯双链 DNA 溶液的吸光系数是 0.020,而变性 DNA 和 RNA 的吸光系数是 0.022
- 1OD A260 dsDNA = 50 μg/ml
- 1OD A260 ssDNA = 33 μg/ml
- 1OD A260 ssRNA = 40 μg/ml
★ DNA/RNA 纯度测定
- OD260/OD280的比值如果在 1.8-2.0 之间,表示所提取的 RNA 纯度较好;如果样品中有蛋白质或酚污染,则 OD260/OD280的比值将明显低于 1.8(因为蛋白质在 280nm 吸收强)。
- 高纯度的 DNA紫外吸收值 260/280 的比值约为 1.8,对于 RNA 其比值约为 2.0。
琼脂糖凝胶电泳检测 RNA
- 由于 RNA 呈单链状态,易形成二级结构而且易降解,因此常在变性条件下进行 RNA 电泳。甲醛是最常用的变性剂,也可用尿素。
- 总 RNA 中 80%-85% 为 rRNA
- rRNA 分子具有确定的大小和核苷酸序列,特别是 28S 和 18S 特征性条带是电泳鉴定总 RNA 纯度和完整性的重要参数。
- 聚丙烯酰胺凝胶电泳主要用来分析小相对分子质量的 RNA, 电泳后如果 rRNA 大小完整,而且 28S rRNA 和 18S rRNA 亮度接近 2:1, mRNA 分布均匀,则认为 RNA 质量较好。
7.2.2 mRNA 的纯化
- 真核细胞的 mRNA 分子最显著的结构特征是具有 5’端帽子结构 (m7G) 和 3’端的 poly(A) 尾巴,这种 poly(A) 结构为真核生物 mRNA 的提取提供了极为方便的选择性标志。
- 实验中常用寡 (dT)-纤维素柱层析法获得高纯度 mRNA。
原理:该方法利用 mRNA 3’末端含有 poly(A) 的特点,当 RNA 流经寡 (dT) 纤维素柱时,在高盐缓冲液的作用下,mRNA 被特异性地结合在柱上,再用低盐溶液或蒸馏水洗脱 mRNA。经过两次寡 (dT) 纤维柱后可得到较高纯度的 mRNA。 - 实验中常用 polyAT Tract mRNA 分离系统 将生物素标记的寡 (dT) 引物与细胞总 RNA 温育,加入与微磁球相连的抗生物素蛋白以结合 polyA mRNA, 通过磁场吸附作用将 poly (A)mRNA 从总 RNA 中分离。

7.2.3 cDNA 的合成
- 真核生物基因组 DNA 庞大,有大量重复序列,很难直接分离得到靶基因片段。而 cDNA 来自反转录的 mRNA,无冗余序列,通过筛选 cDNA 文库,可较快地分离到相关基因。
- 由于 RNA 分子敏感脆弱,在自然状态下难以被扩增,为了研究 mRNA 所包含的功能基因信息,一般将其反转录成稳定的 DNA 双螺旋(cDNA, complementary DNA),再插入到可以自我复制的载体中。
● cDNA 合成过程
- cDNA 的合成包括第一链和第二链 cDNA 的合成
- 第一链 cDNA 的合成是以 mRNA 为模板,反转录成 cDNA, 由反转录酶催化,该酶合成 DNA 时需要引物引导,常用的引物是 oligo(dT)。oligo(dT) 引物一般包含 12~20 个脱氧胸腺密啶核苷酸(dT),后面加一个连接引物(通常为 XhoⅠ等酶切位点)以便于克隆构建。
- 第二链 cDNA 的合成是以第一链为模板,由 DNA 聚合酶催化。常用 RNase H 切割 mRNA-cDNA 杂合链中的 mRNA 序列所产生的小片段为引物合成第二条 cDNA 的片段,再通过 DNA 连接酶的作用连成完整的 DNA 链。
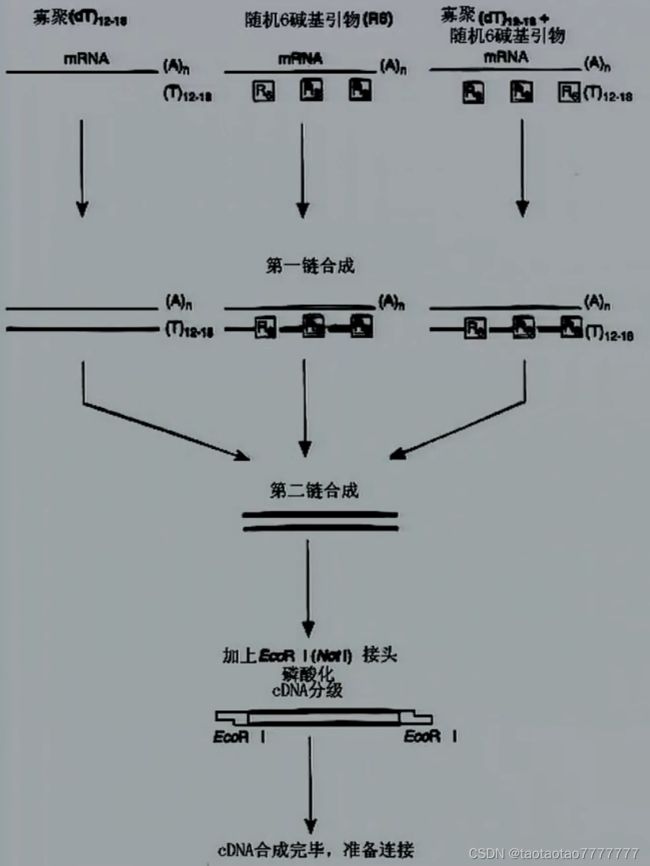
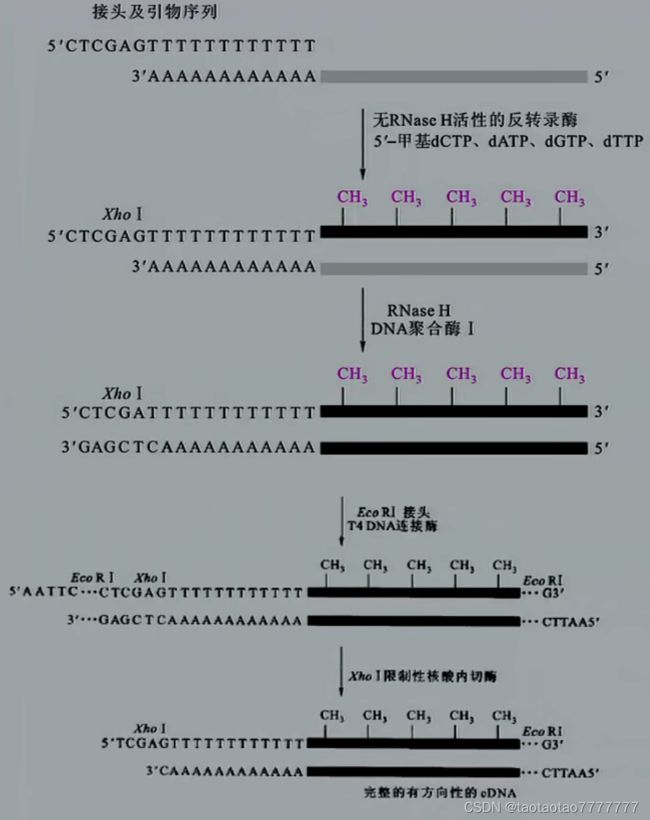
● 定向 cDNA 合成及分子修饰
- 在合成的 cDNA 两端应加上不同内切酶所识别的接头序列,保证所获得双链 cDNA 的方向性。
加入含有另一个酶切位点的黏性接头(如 EcoRⅠ), 与 cDNA 相连接后用 XhoⅠ酶切,使 cDNA 双链 5’端和 3’端分别具有 EcoRⅠ和 XhoⅠ黏端,保证它与载体相连时有方向性。 - 因为绝大多数大肠杆菌细胞都会切除带有 5’-甲基胞密啶 (m5C) 的外源 DNA, 所以实验中常选用 mcrA- mcrB- 菌株以防止 cDNA 被降解。
- 反应体系中一般加入甲基化 dCTP, 保证新合成的 cDNA 链被甲基化修饰,以防止构建克隆时被限制性内切核酸酶切割。
7.2.4 cDNA 文库的构建
- 由于 cDNA 的长度一般为 0.5 ~8 kb, 常用的质粒载体和噬菌体类载体都能满足要求。
- cDNA 文库的载体选择要根据该文库的用途来确定,常用 Uni-zap XR 载体(一种 λ 噬菌粒载体),具备噬菌体的高效性和质粒载体系统可利用蓝白斑筛选的便利,可容纳 0 ~10 kb DNA 插入片段。该载体内部含有 pBluescript 载体的全部序列。
- 重组后可通过体内剪切反应 (in vivo excision) 将 cDNA 插入片段转移到质粒系统中进行筛选、克隆和序列分析。
- 一个比较完整的 cDNA 文库常包含大于 5×105 的独立克隆。
7.2.5 基因文库的筛选
基因文库的筛选是指通过某种特殊方法从基因文库中鉴定出含有所需重组 DNA 分子的特定克隆的过程。- 筛选方法:核酸杂交法、PCR 筛选法、免疫筛选法等。
核酸杂交法
- 核酸杂交法以其广泛的适用性和快速性成为基因文库筛选中最常用的一种方法,常用放射性标记的特异 DNA 探针进行高密度的菌落杂交筛选。

1)将圆形硝酸纤维素膜放在含有琼脂培养基的培养皿表面,将待筛选菌落转移到硝酸纤维素膜上,适当温育。同时保留原来的菌落平板作对照。
2)取出已经长有菌落的膜,用碱液处理,使菌落发生裂解,DNA 随之变性。用蛋白酶 K 处理硝酸纤维素膜去除蛋白质,形成菌落 DNA 的印迹。
3)80℃烘烤滤膜,将 DNA 固定在膜上。将滤膜与放射性标记的 DNA 或 RNA 探针 (针对目的基因设计的探针)杂交,通过放射性自显影显示杂交结果。通过对应于平板上的位置找到相应克隆。
PCR 筛选法
- 前提是已知足够的序列信息并获得基因特异性引物。
- 将整个文库(以质粒或菌落的形式均可)保存在多孔培养板上,用设计好的目的基因探针对每个孔进行 PCR 筛选,鉴定出阳性的孔,把每个阳性孔中的克隆再稀释到次级多孔板中进行 PCR 筛选。重复以上程序,直到鉴定出与目的基因对应的单个克隆为止。
免疫筛选法
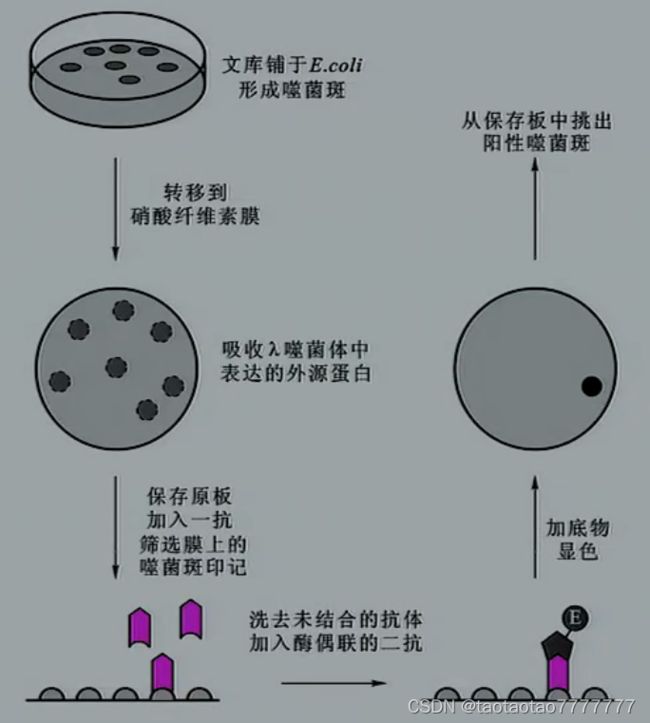
- 该法仅适用于对表达文库(蛋白质水平)的筛选。若实验中靶基因的序列完全未知但是有针对该基因产物的特异性抗体,就可以采用免疫筛选法。
- 免疫检测与菌落或噬菌斑的核酸杂交相似,先将菌落或噬菌斑影印到硝酸纤维素膜上,原位溶解菌落释放抗原蛋白,再用抗体与固定了抗原的膜杂交,抗原抗体结合后,再用标记的二抗与之反应,通过对标记物的检测,就可以找到阳性克隆。
7.3 新基因的发现与克隆
7.3.1 简并引物
- 基于蛋白质序列设计寡核苷酸探针
- 应用:从已知蛋白质或者多肽序列出发鉴定基因的序列
- 参考资料:简并引物设计的方法(含详细步骤)

7.3.2 RACE(cDNA 末端快速扩增法)
- cDNA 末端快速扩增法(apid amplification of cDNA ends,
RACE):在已知 cDNA 序列基础上克隆 5’或 3’端缺失序列的技术。 - 根据已知序列设计基因片段内部特异引物,由该片段向外侧进行 PCR 扩增得到目的序列。
- 用于扩增 5’ 端的方法称为 5’ RACE(巢式 PCR),用于扩增 3’端的称为 3’ RACE。
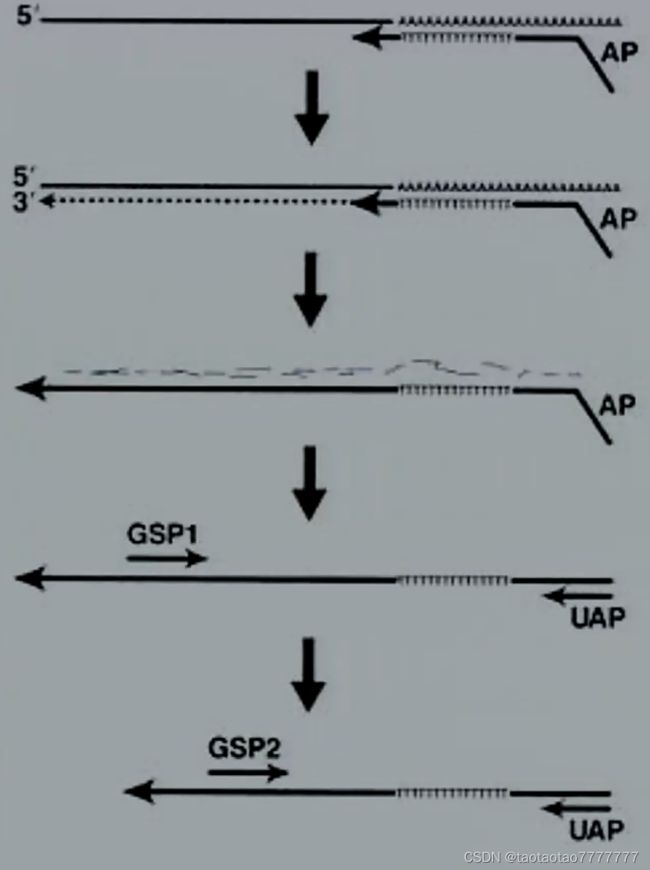
5’ RACE
- 在反转录酶的作用下,用已知基因片段内部特异性引物 (gene-specific primer 1, GSP1) 启始 cDNA 第一条链的合成。
- 用 RNase 混合物降解模板 mRNA,纯化已合成的 cDNA 第一条链。
- 由末端转移酶在已合成的 cDNA 链的 3’末端连续加入 dCTP, 形成 oligo(dC) 尾巴。
- 以连有 oligo(dG) 的锚定引物 AP( anchor primer) 和基因片段内部特异引物 GSP2 进行巢式 PCR(提高特异性)扩增,以期得到目的基因 5’端片段。

3’ RACE
- 在反转录酶的作用下,以连有可以与 mRNA 3’末端配对的 oligo(dT) 锚定引物 AP 启始 cDNA 第一条链的合成。
- 用 RNase 降解模板 mRNA。
- 用通用锚定引物 UAP 和基因片段内部特异引物 GSP 进行巢式 PCR 扩增得到目的基因 3’ 端片段。
7.3.3 RNA 的差异显示技术(DDRT-PCR) (此方法现已不用)
- 应用 mRNA 差别显示技术克隆目的基因。
- 不同基因在生物个体发育的不同阶段或不同的组织、细胞中发生的按时间、空间进行有序表达的方式称为基因的差别表达 (differential expression)。
- 1992 年,美国波士顿 Dena-Farber 癌症研究所的科学家 P.Liang 和 A.D. Pardee 发明了 mRNA 的差别显示(mRNA differential display)技术,简称
DDRT-PCR,可以从一对不同基因型的细胞群体所产生的约 15 000 种 mRNA 中有效地鉴定并分离出差别表达的基因。 - 设计合成了 12 种由 11 个或 12 个连续的脱氧胸苷酸 (dTMP) 加上两个 3’-端锚定脱氧核苷酸(大大提高引物结合力)组成的 3’引物,用以反转录 mRNA,合成第一链 cDNA。每一种人工合成的寡核苷酸引物,都能够把总 mRNA 群体的 1/12 分子反转录成 mRNA-cDNA 杂合分子。
- 从一对不同的组织或器官中分离总 mRNA,分别称为 A 组和 B 组。

7.3.4 cDNA 差示分析法 (RDA)
- cDNA 差示分析法 (representational difference analysis,
RDA) 充分发挥了 PCR 以指数形式扩增双链 DNA 模板,而仅以线性形式扩增单链模板的特性,通过降低 cDNA 群体复杂性和更换 cDNA 两端接头等方法,特异性扩增目的基因片段。 - 4 碱基切割酶处理 Tester(T, 实验材料)和 Driver(D, 探针材料),形成平均为 256 bp 的代表群,保留了绝大部分遗传信息。
- 第一次 T 减 D 反应中两者的浓度比就要求达到 1:100 或更高,经过 2~3 次重复后,T 群体中非特异性序列几乎没有偶然逃脱的可能性。
- 每次 T 减 D 反应后仅设置 72℃复性与延伸、94℃变性这两个参数共 20 个 PCR 循环,PCR 产物的特异性和所得探针的纯度高。
- ① T、D 两组 cDNA 有差异,实验目的是找出差异基因;
② 先用相同方法处理 T、D,加接头,PCR 扩增;
③ 消化掉 T 和 D 的接头后, 给 T 加一个新接头,将 T:D=1:100 混合后,高温打散,再低温随机配对杂交,产生 3 种产物(TD 少量 /TT 极少 /DD 大量)
◆ 由于 D 的量远大于 T,T 与 D 中相同的部分就会与 D 形成杂合体,而只有 T 特异的基因才能重新组合成 TT,不会受 D 的影响。
◆ DD 没有接头不能扩增;TD 链只有一端有接头,以线性形式扩增;特异的 TT 链两端都有接头,以指数形式扩增。
④ 将相对特异的 TT 再与 D 以 1:100 混合扩增,重复 2-3 次,筛选出所有相对于 D 特异的 T 的差异基因。 - 参考资料:几种基因克隆的常用方法介绍(二)

7.3.5 Gateway 大规模克隆技术
- Gateway 技术利用 λ 噬菌体进行位点特异性 DNA 片段重组,实现了不需要传统的酶切联接过程的基因快速克隆和载体间平行转移。
- 包括 TOPO 反应和 LR 反应两步。
TOPO 反应
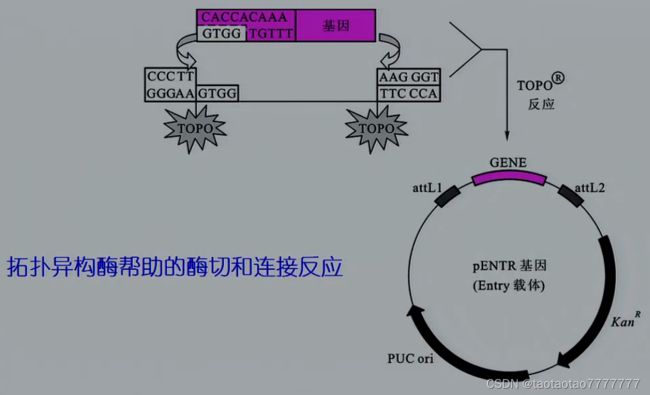
- 将目的基因 PCR 产物连入 Entry 载体。载体上的 CCCTT 被拓扑异构酶所识别并被切割,通过与切口处的磷酸基团与酶形成共价键,将该酶偶联在载体上。
- 载体的 5’GTGG 粘性末端攻击 PCR 产物的互补性末端并与接头序列 CACC(需要在目的基因引物前面加 CACC 接头) 退火,使 PCR 产物以正确方向连入 Entry 载体。
- 试剂盒提供的线性质粒的 3’端都带有一个拓扑异构酶
LR 反应
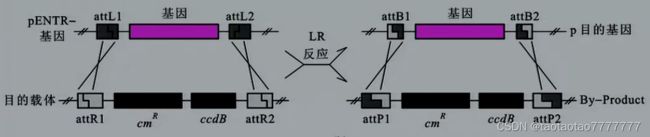
- 将目的片段从 Entry 载体中重组入表达载体。Entry 载体上基因两端具有 attL1 和 attL2 位点,目的载体上含有 attR1 和 attR2 位点,在重组蛋白的作用下发生定向重组(同源重组),形成新的位点 attB1 和 attB2,将目的基因转移到表达载体中。
● Gateway 克隆试剂盒
- 一般会提供至少两种载体质粒:Doner 载体、Destination 载体。
- 先把目的片段转到 Donor 载体上形成 Entry 载体,Entry 载体把目的片段换到 Destination 载体(序列特异性重组)上变成最终的克隆产物。
- 要求被置换目的片段两端必须有特定序列,因此设计扩增目的片段的引物时要在目的片段两端加 20bp 特定序列,引物设计方案在武剂盒说明中有详细介绍。
- 参考资料(b 站):分子克隆方法| Golden Gate/ Gateway/ Infusion
7.3.6 基因的图位克隆法 (map-based cloning)
- 所有具有某种表型的基因都可以通过该方法克隆得到。
- 首先,通过构建遗传连锁图,将目的基因定位到某个染色体的特定位点,并在其两侧确定紧密连锁的 RFLP 或 RAPD 分子标记。
◆ 构建遗传连锁图:把不同的染色体构建到不同载体上,找到影响表型变化的那条染色体;将该条染色体切一半,以此类推,不断缩小染色体范围,从而定位到表型相关的特定染色体位点。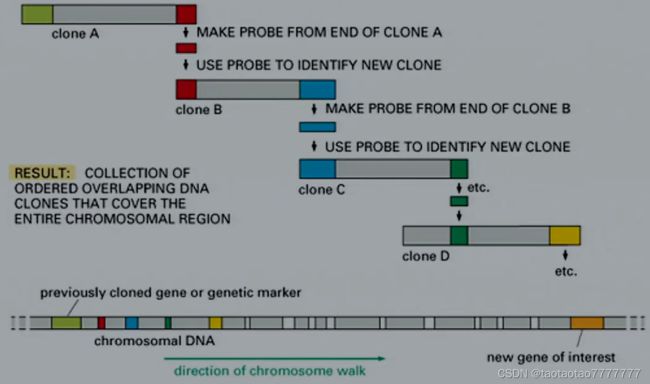
- 其次,通过对许多不同生态型个体的大量限制性内切核酸酶和杂交探针的分析,找出与目的基因距离最近的分子标记,通过染色体步移技术将位于这两个标记之间的基因片段分离并克隆出来,再根据基因功能互作原理鉴定目的基因。
◆ 染色体步移法克隆基因

- 在 RFLP 作图中,
连锁距离是根据重组率来计算的,1 cM(厘摩)相当于 1% 的重组率。
人类基因组中,1 cM = 1 000 kb; 拟南芥中,1 cM ≈ 290 kb; 小麦中,1 cM ≈ 3500 kb.
用图位克隆法获得水稻脆杆基因 BCL

- A. 将 BCL 定位于水稻 3 号染色体 (Chr3)分子标记 C524a 和 RM16 之间;
- B. 覆盖 BCL 位点的 BAC 大片段。
- C. BCL 位点的精细定位。BCL 位点被定位于分子标记 P2 和 P4 之间与 P3 共分离。
- D. BCL 基因结构。红色为编码区,白色为 5 和 3’非翻译区,黑色线段为内含子。bcl-1 和 bcl-2 表示两个突变位点。
7.3.7 反向 PCR 技术
常规 PCR 技术能扩增两引物之间的 DNA 区段,然而,有时我们也希望扩增位于靶 DNA 区段之外的未知 DNA 序列,这就需要应用反向 PCR (reverse PCR)技术。
反向 PCR 的基本操作程序
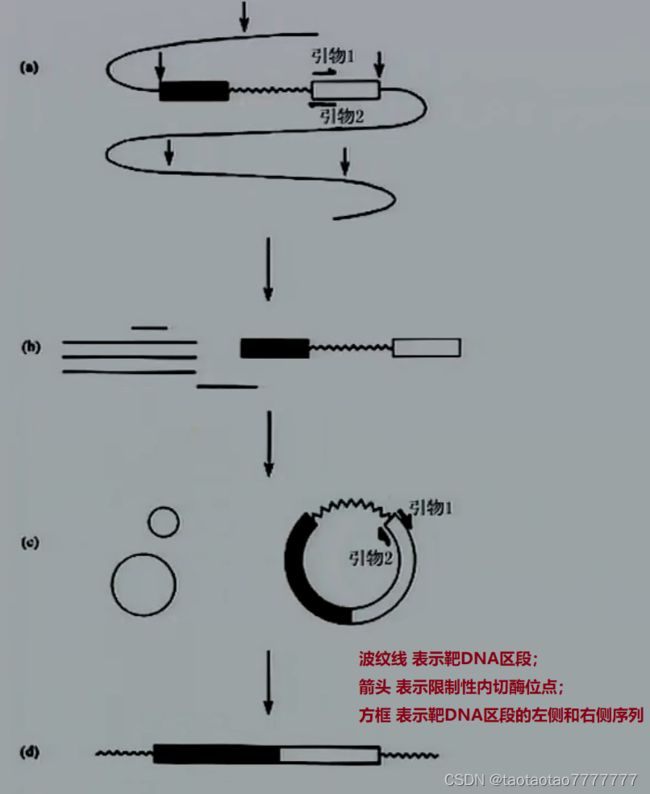
- 用一种在靶序列(已知序列)上没有酶切位点的核酸限制性内切酶消化 DNA;
- 产生大小不同的线性 DNA 片段群体,其中靶 DNA 区段的 DNA 分子长度不超过 2~3kb,经连接后重新环化;
- 按靶序列设计的一对引物与互补序列退火结合,其延伸方向如箭头所指。
- 参考资料:SnapGene 操作指南 | 反向 PCR
7.3.8 TAIL-PCR 克隆 T-DNA 插入位点侧翼序列
- 热不对称交错多聚酶链式反应 (thermal asymmetric inter-laced PCR,
TAIL-PCR) 常用于扩增 T-DNA 插入位点侧翼序列,从而获得转基因植物插入位点特异性分子证据。 - TAIL-PCR 使用一套巢式特异引物(T-DNA 边界引物,
TR)和一个短的随机简并引物 (AD)。 T-DNA:转移 DNA (Transfer DNA), 也叫三螺旋 DNA,是指一种由三股 ssDNA 旋转螺旋形成的一种特殊结构。农杆菌 Ti(tumor inducing) 或 Ri(root inducing) 质粒中的一段 DNA 序列,可以从农杆菌中转移并稳定整合到植物核基因组中。- 已知 T-DNA 插入了基因组,想知道 T-DNA 下游的序列
TAIL-PCR 流程
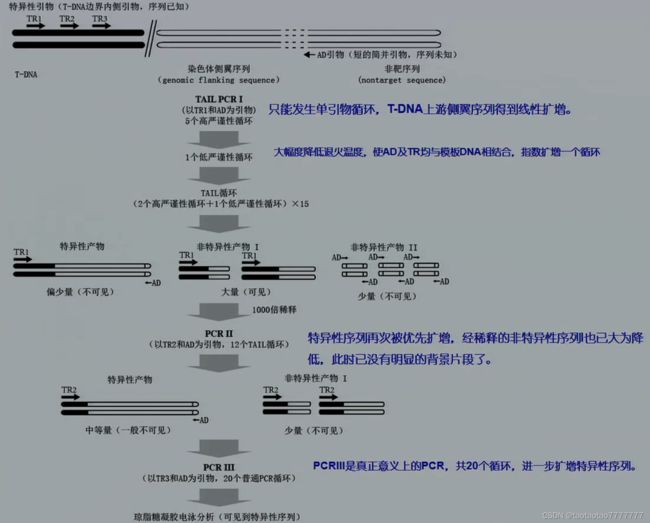
- 第一轮反应 (primary reaction,
PCRⅠ) :是 TAIL-PCR 的重要环节
① 先进行 5 个高严谨性循环,特异性引物 TR1 与模板退火(只能发生单引物循环,T-DNA 上游侧翼序列得到线性扩增)。
② 再大幅度降低退火温度,使 AD 及 TR1 均与模板 DNA 相结合,指数扩增 1 个循环。
③ 此后,2 个高严谨 + 1 个低严谨循环交替进行,共 15 个循环。
◆ 结果: 特异性序列(两端分别拥有 TR1 和 AD 序列,偏少量,不可见)和非特异性序列Ⅰ(只有 TR1, 没有 AD 序列,大量)大大超过非特异性序列Ⅱ(两端均为 AD 序列,少量,不可见)。 PCRⅡ以 TR2 为特异性引物与 AD 配对,进行 12 个 TAIL-PCR 循环,特异性序列再次被优先扩增,非特异性序列Ⅰ也大大降低,此时已没有明显的背景片段。PCRⅢ是真正意义上的 PCR,用 TR3 为特异性引物与 AD 配对,共 20 个循环,进一步扩增特异性序列。
本讲总结:
① 核酸的凝胶电泳;
② 细菌转化;
③ 感受态细胞;
④ 聚合酶链式反应(PCR)技术;
⑤ 实时定量 PCR, TaqMan 探针;
⑥ 重亚硫酸盐测序技术;
⑦ 基因组 DNA 文库构建,cDNA 文库;
⑧ RNA 浓度的测定;mRNA 分离;
⑨文库的三种筛选方法;
⑩ 新基因发现:
简并引物 RACE (Rapid amplification of cDNA ends);
RNA 的差异显示 (mRNA differential display);
cDNA 差示分析法 (RDA, Representation Difference Analysis);
基因的图位克隆法 (map-based cloning);
反向 PCR;
TAIL-PCR