GO和kEGG富集分析
文章目录
- 前言
- 一、GO和KEGG
-
- 1. **GO 富集分析:**
- 2. KEGG 富集分析:
- 二、使用步骤
-
- 1.数据处理
- 2. GO分析
- 3.KEGG富集
- 总结
前言
GO(Gene Ontology,基因本体)富集和 KEGG(Kyoto Encyclopedia of Genes and Genomes,京都基因与基因组百科全书)富集分析能够从不同角度揭示基因的功能和生物学意义
一、GO和KEGG
1. GO 富集分析:
- 说明基因在分子功能(Molecular Function)、生物过程(Biological Process)和细胞组成(Cellular Component)三个方面的特征和倾向。
- 帮助了解基因参与的具体生物学活动,例如基因是具有催化活性、结合能力,还是参与细胞分裂、信号转导等过程,以及在细胞的哪个部位发挥作用。
2. KEGG 富集分析:
- 反映基因所参与的代谢通路、信号转导通路、疾病相关通路等。
- 揭示基因在细胞整体的生化反应和生理过程中的协同作用和调控关系。
- 有助于理解基因在疾病发生发展、药物作用机制等方面的作用。
综合来看,GO 富集更侧重于基因功能的分类描述,而 KEGG 富集则更侧重于基因在生物系统中的通路级别的相互作用和调控。两者结合可以更全面深入地理解基因的功能和在生物学系统中的角色
二、使用步骤
1.数据处理
library(tidyverse)
library(ggplot2)
library(ggrepel)
library(readxl)
setwd('G:/R/TCGA/venn')
# 这里的excel文档是之前TCGA差异分析得到的数据,主要用longFC,p值,FDR和基因名
COVIN_24_wpi_DEGs <- read_xlsx('long_CONVID_19_deg.xlsx',sheet='24 wpi DEGs')
COVIN_24_wpi_DEGs <- data.frame(COVIN_24_wpi_DEGs$logFC,COVIN_24_wpi_DEGs$PValue,COVIN_24_wpi_DEGs$FDR,COVIN_24_wpi_DEGs$external_gene_name,COVIN_24_wpi_DEGs$ensembl_gene_id,COVIN_24_wpi_DEGs$description,row.names = COVIN_24_wpi_DEGs$external_gene_name)
colnames(COVIN_24_wpi_DEGs) <- c('logFC','FDR','PValue','genename','gene_id','description')
COVIN_24_wpi_DEGs$time <- c('24 w.p.i.')
common_difference_gene_24_wpi <- read.csv('common_difference_gene_24_wpi.csv',row.names = 1)
selected_data_24_wpi <- COVIN_24_wpi_DEGs[COVIN_24_wpi_DEGs$genename %in% t(common_difference_gene_24_wpi), ]
threshold <- 0.3
selected_data_24_wpi$regulation <- ifelse(selected_data_24_wpi$logFC > threshold, "Up-Regulated", ifelse(selected_data_24_wpi$logFC < -threshold, "Down-Regulated", ifelse(abs(selected_data_24_wpi$logFC) < threshold, "NotSig", "Unknown")))
# 获取上下调基因
top <- 100
top_genes_24_wpi <- bind_rows(
selected_data_24_wpi %>%
filter(regulation == 'Up-Regulated') %>%
arrange(desc(abs(logFC))) %>%
head(top)
)
bottom_genes_24_wpi <- bind_rows(
selected_data_24_wpi %>%
filter(regulation == 'Down-Regulated') %>%
arrange(desc(abs(logFC))) %>%
head(top)
)
# 保存数据之后方便之后的通路分析
library(openxlsx)
dir.create('differential gene/result/24_wpi/data_100')
write.xlsx(selected_data_24_wpi,'./differential gene/result/24_wpi/data_100/data_24_wpi.xlsx',rowNames = TRUE)
write.xlsx(top_genes_24_wpi,'./differential gene/result/24_wpi/data_100/top_genes_24_wpi.xlsx',rowNames = TRUE)
write.xlsx(bottom_genes_24_wpi,'./differential gene/result/24_wpi/data_100/bottom_genes_24_wpi.xlsx',rowNames = TRUE)
2. GO分析
setwd('G:/R/TCGA/venn/differential gene')
library(clusterProfiler)
# 这里org.Hs.eg.db需要通过BiocManager包进行下载,如果多次尝试下载不成功的恶化,可以先更改镜像源,之后再尝试下载
# options(BioC_mirror="https://mirrors.tuna.tsinghua.edu.cn/bioconductor")
# BiocManager::install('org.Hs.eg.db')
library(org.Hs.eg.db)
library(ggplot2)
library(readxl)
# 读取数据
upgene <- read_xlsx('./result/24_wpi/data_100/up_genes_24_wpi.xlsx')
rownames(upgene) <- upgene$...1
Genes <- bitr(rownames(upgene),
fromType = 'SYMBOL',
toType = c('ENTREZID'),
OrgDb = org.Hs.eg.db)
# GO富集分析和结果存储
GO <- enrichGO(gene = Genes$ENTREZID, # 输入基因的ENTREZID
OrgDb = org.Hs.eg.db, # 注释信息
keyType = 'ENTREZID',
ont = 'ALL',# 可选条目BP/CC/MF
pAdjustMethod = 'BH',# p值的校正方式
qvalueCutoff = 1,# q的阈值
pvalueCutoff = 1, # p的阈值
minGSSize = 5,
maxGSSize = 500,
readable = TRUE # 是否将ENTREZID转换为symbol
)
dir.create('./result/24_wpi/up_100')
write.csv(data.frame(ID=rownames(GO@result),GO@result),'./result/24_wpi/up_100/up_genes_24_wpi_GO.csv')
# 柱状图
pdf(file = './result/24_wpi/up_100/up_genes_24_wpi_GO_barplot.pdf',width = 10,height = 8)
barplot(GO,drop=TRUE,
showCategory = 6,
split='ONTOLOGY') +
facet_grid(ONTOLOGY~.,scales = 'free')
dev.off()
# 点图
pdf(file = './result/24_wpi/up_100/up_genes_24_wpi_GO_dotplot.pdf',width = 10,height = 8)
dotplot(GO,showCategory=6, split='ONTOLOGY')+
facet_grid(ONTOLOGY~.,scales = 'free')
dev.off()
# 不分面
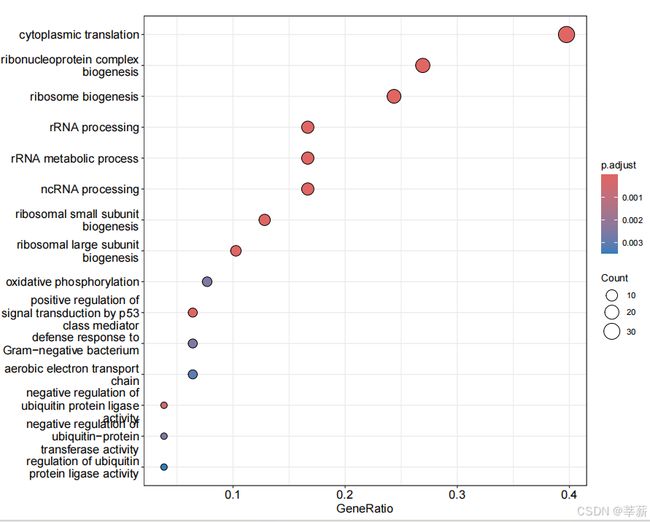
pdf(file = './result/24_wpi/up_100/up_genes_24_wpi_GO_dotplot_nosplit.pdf',width = 10,height = 8)
dotplot(GO,
x='GeneRatio',
color='p.adjust',
showCategory=15,
size=NULL,
split=NULL,
font.size=13,
title='',
orderBy='x',
label_format=30)
dev.off()
3.KEGG富集
KEGG <- enrichKEGG(Genes$ENTREZID,
organism = 'hsa',
keyType = 'kegg',# kegg数据库
pAdjustMethod = 'BH',
pvalueCutoff = 1,
qvalueCutoff = 1)
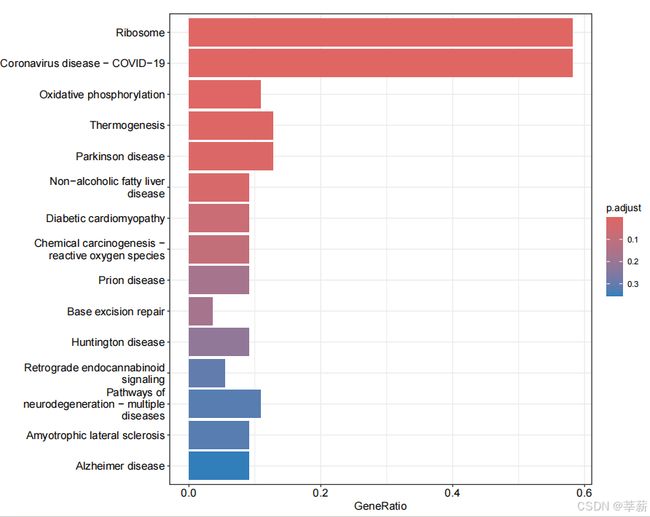
pdf(file = './result/24_wpi/up_100/up_genes_24_wpi_KEGG.pdf',width = 10,height = 8)
barplot(KEGG,
x='GeneRatio',
color='p.adjust',
showCategory=15)
dev.off()
pdf(file = './result/24_wpi/up_100/up_genes_24_wpi_KEGG_dotplot.pdf',width = 10,height = 8)
dotplot(KEGG)
dev.off()
# View(as.data.frame(KEGG)) # 以数据框的形式展示KEGG的结果
# browseKEGG(KEGG,'hsa03010') # 打开KEGG官网查看相关通路的信息
# 结果存储
KEGG_results <- DOSE::setReadable(KEGG,
OrgDb = org.Hs.eg.db,
keyType = 'ENTREZID')
write.csv(data.frame(ID=rownames(GO@result),GO@result),'./result/24_wpi/up_100/up_genes_24_wpi_KEGG.csv')
总结
图片很多,这里就不再一一展示了,有兴趣的朋友可以自己常以跑一下,了解一下它们之间的区别,需要数据的话私我领取!!!