QIIME2进阶三_用QIIME2实现对数据的质量控制
本文主要介绍了使用生物信息软件QIIME2中的DADA2与Deblur插件对扩增子基因序列进行质量控制。
本教程将使用来自人源化(humanized)小鼠的一组粪便样品,展示16S rRNA基因扩增子数据的“典型”QIIME 2分析。本教程旨在探讨人源化小鼠的遗传背景影响微生物群落的假设。然而,我们还需要考虑其他可能驱动微生物结构而不是小鼠基因型的混杂因素。
序列质量控制和特征表 Sequence quality control and feature table
QIIME 2插件多种质量控制并生成特征表的方式主要有两种,一种是通过去噪,即生成扩增/绝对序列变体(Absolute Sequence Variants,ASV),ASV是最近发展的新一代方法,在功能上提供更好的分辨率。ASV可以基于400bp或更多序列中单个核苷酸的差异来分离特征,甚至超过99%同一性OTU聚类的分辨率。目前在QIIME 2 中可通过DADA2(q2-dada2)和Deblur(q2-deblur)插件实现。第二种是通过聚类生成操作分类单元(Operational Taxonomic Units,OTU),这种方法自2010年以来便得到了广泛应用。QIIME 2目前可通过q2-vsearch插件实现。两种方法不推荐组合使用。本教程将着重介绍DADA2和Deblur两种方法。
OTU(Operational Taxonomic Units):是通过一定距离计算两两不同序列之间的距离度量和相似性,设置特定的阈值,获得同一阈值下的距离矩阵,进行聚类操作,形成不同的分类单元。
方法一:DADA2
二代测序的错误是随机发生的(即,任意两条序列的测序错误相对是随机发生的,一条序列的任意两个位置的测序错误也是随机发生的,不存在关联性)。DADA2质量控制过程将过滤掉在测序数据中鉴定的任何phiX序列(最主要的目的:1、调节碱基平衡,改善测序仪的空间校正,便于后期提高base calling的准确性;2、由于Phix序列已知基因组较小,在测序的过程中Illumina的测序仪就开始将测的read与phix基因组进行比较,预估测序指标。)通常存在于标记基因Illumina测序数据中,用于提高扩增子测序质量,并同时过滤嵌合序列(即嵌合基因,就是两个基因共用一段DNA序列,这两个基因称为嵌合基因)。在DADA2中,双端合并,去除嵌合体,截去接头序列降噪生成feature table都是一步完成的。所以,运行DADA2之前要确保测序数据满足以下规范:
(1)样品已被拆分好,即每个样品一个fq/fastq文件(或者双端成对fq文件);
(2)已经去除非生物核酸序列,比如:引物(primers),接头(adapters or barcodes),linker等;
(3)如果样品是下机的双端测序,其应具有双端测序的相匹配的两个fq文件。
使用DADA2插件进行质量控制:
time qiime dada2 denoise-single
--i-demultiplexed-seqs demux_seqs.qza
--p-trunc-len 150
--o-table dada2_table.qza
--o-representative-sequences dada2_rep_set.qza
--o-denoising-stats dada2_stats.qza
参数解读
--p-trim-left:截取左端低质量序列。用于切除低质量序列、barocde或引物。
--p-trunc-len:序列截取长度,也是为了切除有段低质量序列。一般从序列质量开始大幅度下降的位置开始切除。
| 命令注释: (1)在使用qiime dada2 denoise-single/ qiime dada2 denoise-paired时可设置--p-n-threads 参数,用于设置运行时使用的线程数量。线程越多,则运行速度越快。当线程设置为0时则默认使用全部线程; (2)--p-trim-left截取左端低质量序列,有时用于切除低质量序列、barocde或引物。查看demux_seq.qzv文件中的箱线图,左端质量都很高,无低质量区,设置为0;或可直接忽略此参数设置; (3)--p-trunc-len序列截取长度,也是为了去除右端低质量序列,我们看到大于150以后,质量下降极大,甚至中位数都下降至20以下,需要全部去除,综合考虑决定设置为150; (4)当处理双端数据时,需考虑截取后的序列是否可以成功拼接。目前最短的拼接长度为引物长度+12bp。 |
生成统计结果:
qiime metadata tabulate
--m-input-file dada2_stats.qza
--o-visualization dada2_stats.qzv

内容为每个样本的输入、过滤、去噪和非嵌合体的统计结果。展示了样本的质量控制结果,用于样本异常筛选和特征表抽平标准化。
生成特征表摘要:
qiime feature-table summarize
--i-table dada2_table.qza
--o-visualization dada2_table.qzv
--m-sample-metadata-file metadata.tsv
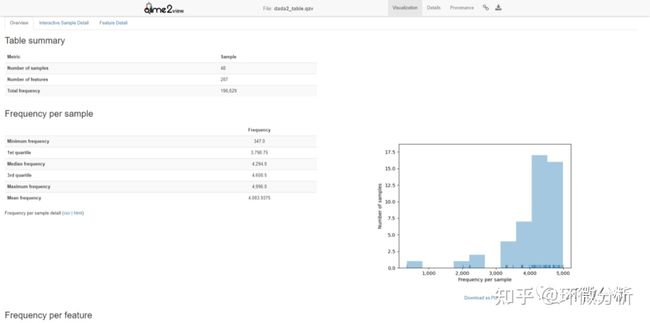
上图为特征表摘要,展示了样本数、特征数和分布等信息。

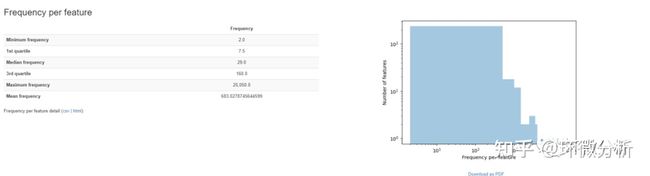
上图展示了每个样本中的特征数目。

上图为特征信息,分别为特征名称、出现频率和共出现在多少个样本中。
生成代表序列摘要:
qiime feature-table tabulate-seqs
--i-data dada2_rep_set qza
--o-visualization dada2_rep_set.qzv

上图为代表序列统计信息。有长度摘要、不同百分比下的长度统计和具体代表序列碱基信息。
方法二:Deblur
PCR和测序过程中的噪音限制了区分更相近的物种。一些特殊的生态应用与科学研究需要更精确的物种区分。因此,提出了Deblur去噪的方法。Deblur方法提出了sub-operational-taxonomic-unit (sOTU) 提出更精确的分类亚OTU的概念,此概念与ASV意义相同,只是名字不同。
Deblur具有以下特点:
(1)使用误差分布来获得假定的无误差序列;
(2)减少了计算的需求,得到了更高的特异性和敏感性;
(3)只受扩增序列读长和多样性的限制;
(4)可以在单个样本水平上使用。
按测序碱基质量过滤序列
time qiime quality-filter q-score
--i-demux demux_seqs.qza
--o-filtered-sequences demux-filtered.qza
--o-filter-stats demux-filter-stats.qza
输出结果文件:
demux-filtered.qza: 序列质量过滤后结果;
demux-filter-stats.qza: 序列质量过滤后结果统计。
deblur去噪16S过程,输入文件为质控后的序列,设置截取长度参数,生成结果文件有代表序列、特征表、样本统计:
time qiime deblur denoise-16S
--i-demultiplexed-seqs demux-filtered.qza
--p-trim-length 150
--o-representative-sequences rep-seqs-deblur.qza
--o-table deblur-table.qza
--p-sample-stats
--o-stats deblur-stats.qza
可视化输出文件:
time qiime metadata tabulate
--m-input-file demux-filter-stats.qza
--o-visualization demux-filter-stats.qzv
time qiime deblur visualize-stats
--i-deblur-stats deblur-stats.qza
--o-visualization deblur-stats.qzv
time qiime feature-table tabulate-seqs
--i-data rep-seqs-deblur qza
--o-visualization rep-seqs-deblur.qzv
time qiime feature-table summarize
--i-table deblur-table.qza
--o-visualization deblur-table.qzv
--m-sample-metadata-file metadata.tsv
如果使用deblur-16S,deblur执行初始的正向过滤步骤,其中它丢弃与85% GreenGenes 数据库中OTU的序列小于60%相似性的任何序列。如果不想执行此步骤,请使用deblur-other方法。
deblur目前只能对单端序列进行去噪。如果提供末合并的双端序列为输入,将对反向序列不作任何操作。请注意,deblur接受合并的序列,并将它们视为单端序列,因此如果使用deblur进行去噪,需要先合并读取。
本文提供所有输出结果文件,百度网盘下载链接:
https://pan.baidu.com/s/1uj-QSjzxS3mRrgj_vWFaAw
提取码:1234
这篇推文对你有帮助吗?喜欢这篇文章吗?喜欢就不要错过呀,关注本知乎号查看更多的环境微生物生信分析相关文章。亦可以用微信扫描下方二维码关注“环微分析”微信公众号,小编在里面载入了更加完善的学习资料供广大生信分析研究者爱好者参考学习,也希望读者们发现错误后予以指出,小编愿与诸君共同进步!!!

学习环境微生物分析,关注“环微分析”公众号,持续更新,开源免费,敬请关注!
转载自原创文章:
QIIME2进阶三_用QIIME2实现对数据的质量控制
最后,再次感谢你阅读本篇文章,真心希望对你有所帮助。感谢!