GEMMA 全基因组关联分析+CMplot多性状曼哈顿+QQ图脚本
这里写自定义目录标题
- GEMMA 全基因组关联分析+CMplot多性状曼哈顿+QQ图脚本
GEMMA 全基因组关联分析+CMplot多性状曼哈顿+QQ图脚本
###GEMMA 全基因组关联分析+CMplot多性状曼哈顿+QQ图脚本
#作者:刘济铭
##########################
GWAS理论和基本结果理解已经有很多大神进行了解读,因此不再赘述,想要学习请移步知乎或各大基因课平台学习。本人发现目前缺乏具体实用脚本,因此本帖只提供本人脚本供大家交流学习,本人脚本不是最好肯定也会存在问题,请大家见谅。
###全基因组关联分析前需对SNP文件利用repeatmodeler和repeatmasker去除重复片段,后续分析均采用去重复片段SNP文件分析。
#vcftools 过滤VCF文件
vcftools --vcf testnorepeats.recode.vcf --min-alleles 2 --max-alleles 2 --maf 0.05 --max-missing 0.3 --minQ 20 --recode --out test
#此步骤根据各自需求决定,去除多余vcf文件中多余个体数据,id.txt自行构建,个体名按列排布即可
vcftools --vcf test.recode.vcf --recode --recode-INFO-all --stdout --remove id.txt > out.vcf
#转plink bed,ped文件
vcftools --vcf out.vcf --plink --out testnorepeat
plink --file testnorepeat --make-bed --out testnorepeat --noweb
## 下载GEMMA
wget -c https://github.com/genetics-statistics/GEMMA/releases/download/0.98.1/gemma-0.98.1-linux-static.gz
## 解压
gzip -d gemma-0.98.1-linux-static.gz
## 设置权限
chmod a+x gemma-0.98.1-linux-static
##!!!!!计算亲缘关系矩阵前必须将plink所得fam文件第六列及以后添加各个体相对应表型数据,后续关联分析用-n定位哪个表型,-n 1 为第六列表型分析,-n 2 为第七列表型分析,以此类推
##运行gemma
####计算亲缘关系矩阵,-bfile:输入Plink二进制格式文件的前缀。-gk:指定生成的kinship矩阵类型。
#-gk 1 为centered matrix,-gk 2 为standardized matrix。
#-o:输出文件前缀。
./gemma-0.98.1-linux-static -bfile testnorepeat -gk 2 -o kinship
#运行gemma关联分析,可利用-a输入参考基因组基因注释gff3文件
./gemma-0.98.1-linux-static -bfile testnorepeat -k ./output/kinship.sXX.txt -n 39 -a final.gene.gff3 -lmm 4 -o out
#完成关联分析
#利用CMplot R包实现曼哈顿图和qq图制作
##首先在linux系统中提取gwas分析结果文件中1,2,3,13列数据
cut -f 1,2,3,13 out.assoc.txt > out1.assoc.txt
#对换第1列和第二列位置
awk '{print $2,$1,$3,$4}' out1.assoc.txt > out2.assoc.txt
###利用vim工具更改表头为SNP Chromosome Position Trait1 Trait2 Trait3。。。。。。。
###此时out2.assoc.txt为R CMplot作图输入文件
#### 计算GWAS显著关联SNP位点阈值,此处采用Bonferroni correction = 显著性水平(0.01/0.05)/ Me Me为去冗余独立SNP数量,可使用plink --file testnorepeat --r 指令得到,也可自行设定阈值范围
###!!!以下阈值为本人数据计算结果,大家请自行根据数据计算更改后使用
cat out.assoc.txt | awk -F ' ' '$13<5.78e-09{print $0}' > out.top1.out
cat out.assoc.txt | awk -F ' ' '$13<2.89e-08{print $0}' > out.top5.out
###提取SNP位置信息
awk '{print $1"\t"$3}' out.top1.out > out1.snp.position
awk '{print $1"\t"$3}' out.top5.out > out5.snp.position
###提取SNP上下游各50kb范围,范围可根据各自数据LD或自行参考文献确定
awk 'BEGIN{FS=OFS="\t"}{if(NR>1){ print $1,$2-50000,$2+50000 }}' out1.snp.position | sort -k 1,1 -k 2,2n > out1.snp.bed
awk 'BEGIN{FS=OFS="\t"}{if(NR>1){ print $1,$2-50000,$2+50000 }}' out5.snp.position | sort -k 1,1 -k 2,2n > out5.snp.bed
#####利用bedtools merge工具实现位置重叠SNP融合
bedtools merge -i out1.snp.bed > out1.snpmerged.bed
bedtools merge -i out5.snp.bed > out5.snpmerged.bed
###注意,所得out1.snpmerged.bed可能存在染色体名称与参考基因组注释文件差异,务必统一为参考基因组注释文件染色体名称形式,不然无法获得交叉对比结果
#awk '{print $1"\t"$4"\t"$5"\t"$9}' gene.gff3 > gene_after.gff3
###利用bedtools intersect工具实现显著SNP位点上下游50kb内基因交叉对比提取
bedtools intersect -a /scratch/n2007039f/output/gene.gff3 -b out1.snpmerged.bed -wa > gene_top1.out
bedtools intersect -a /scratch/n2007039f/output/gene.gff3 -b out5.snpmerged.bed -wa > gene_top5.out
####检查数据,查看第13列从小到大情况
###less out.assoc.txt|sort -k 13g|less
####R中CMplot包实现GWASQQ图,曼哈顿图等的单个和多个表型联合作图multi_plot
cut -f 13 out.assoc.txt > LAD
paste FHD FLD.txt FVD.txt > 1
######better to set all Tab to Space
sed 's/\t/ /g' 1 > 2
#使用R作图,建议服务器中使用,计算容量较大
## 加载R包
library("CMplot")
## 导入数据
data <- read.table("out2.assoc.txt",sep=" ",header=T)
#SNP-density plot
CMplot(data,type="p",plot.type="d",bin.size=1e6,chr.den.col=c("darkgreen", "yellow", "red"),file="jpg",memo="",dpi=300,
main="illumilla_60K",file.output=TRUE,verbose=TRUE,width=9,height=6)
## 绘制单个表型Q-Q plot
CMplot(data,plot.type="q",conf.int=TRUE,box=FALSE,file="jpg",memo="",dpi=300,file.output=TRUE,verbose=TRUE,width=5,height=5)
## 绘制单个表型Rectangular-Manhattan plot,注意threshold设置,不同数据集不同阈值设置
CMplot(data,plot.type="m",LOG10=TRUE,threshold=5.78e-09,file="tif",memo="",dpi=500,file.output=TRUE,verbose=TRUE,width=18,height=8,signal.col=NULL)
CMplot(data,plot.type="m",LOG10=TRUE,threshold=2.89e-08,file="tif",memo="",dpi=500,file.output=TRUE,verbose=TRUE,width=18,height=8,signal.col=NULL)
#Genome-wide association study(GWAS)多表型作图,注意threshold设置,不同数据集不同阈值设置
CMplot(data,type="p",plot.type="c",chr.labels=paste("Chr",c(1:14),sep=""),r=0.4,cir.legend=TRUE,
outward=FALSE,cir.legend.col="black",cir.chr.h=1.3,chr.den.col="black",file="jpg",
memo="",dpi=300,file.output=TRUE,verbose=TRUE,width=10,height=10)
CMplot(data,type="p",plot.type="c",r=0.4,col=c("grey30","grey60"),chr.labels=paste("Chr",c(1:14),sep=""),
threshold=c(5.78e-09,2.89e-08),cir.chr.h=1.5,amplify=TRUE,threshold.lty=c(1,2),threshold.col=c("red",
"blue"),signal.line=1,signal.col=c("red","green"),chr.den.col=c("darkgreen","yellow","red"),
bin.size=1e6,outward=FALSE,file="jpg",memo="",dpi=300,file.output=TRUE,verbose=TRUE,width=10,height=10)
####manhatton plot
CMplot(data, plot.type="m", LOG10=TRUE, ylim=NULL, threshold=c(5.78e-09,2.89e-08),threshold.lty=c(1,2),
threshold.lwd=c(1,1), threshold.col=c("black","grey"), amplify=TRUE,bin.size=1e6,
chr.den.col=c("darkgreen", "yellow", "red"),signal.col=c("red","green"),signal.cex=c(1.5,1.5),
signal.pch=c(19,19),file="jpg",memo="",dpi=300,file.output=TRUE,verbose=TRUE,
width=14,height=6)
#####Multi_tracks Q-Q plot
CMplot(data,plot.type="q",box=FALSE,file="jpg",memo="",dpi=300,
conf.int=TRUE,conf.int.col=NULL,threshold.col="red",threshold.lty=2,
file.output=TRUE,verbose=TRUE,width=5,height=5)
部分成图结果展示:
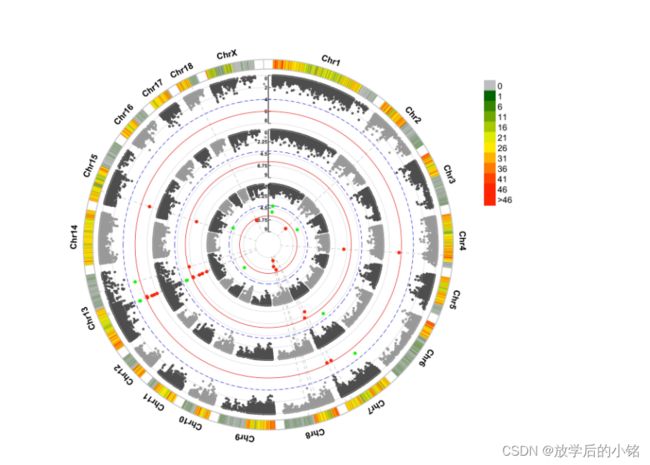

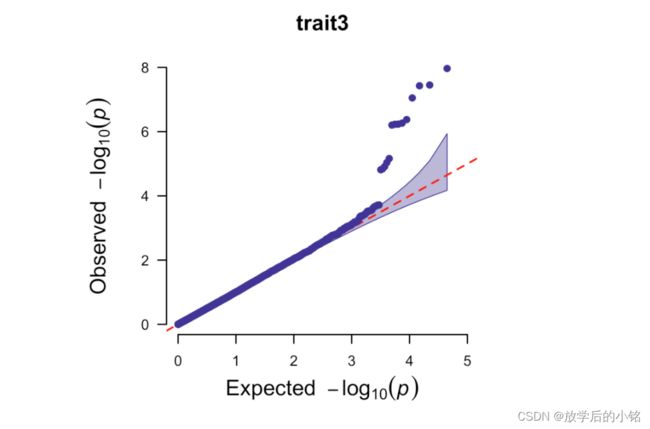
CMplot包参考学习来源:https://github.com/YinLiLin/CMplot,感谢Lilin Yin大神的CMplot包。
CMplot包Author: Lilin Yin
Contact: [email protected]
QQ group: 166305848
Institution: Huazhong agricultural university