分子骨架跃迁工具3DLinker 评测
分子骨架跃迁工具3DLinker来源于文章:

文章链接:3DLinker: An E(3) Equivariant Variational Autoencoder for Molecular Linker Design (arxiv.org)
与之前介绍的骨架跃迁工具不同,3DLinker是变分自动编码器模型,可以在分子linker 设计同时生成分子和分子的坐标。文章引入了空间归纳偏差:equivariance E(3) transformations (等方差E3变换)。
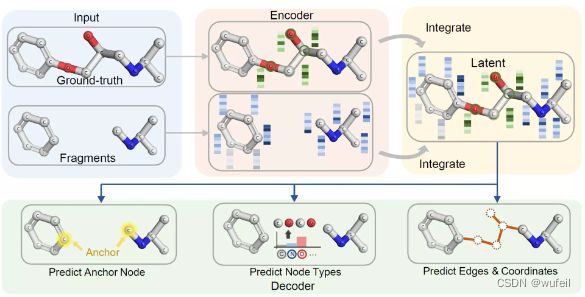
3DLinker以两个分子片段作为输入,输出中间的Linker部分,在输出分子结构的同时还输出linker的坐标。
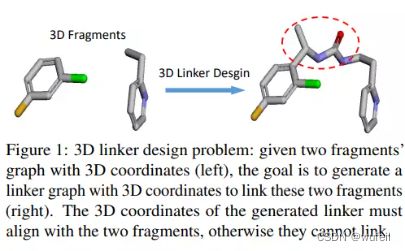
本文对3DLinker进行骨架跃迁进行简单测评。
1.下载源码
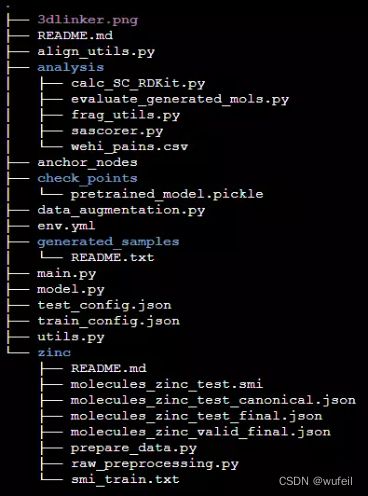
git clone https://github.com/YinanHuang/3DLinker.git项目结构如下:
2.安装环境
ps: pytorch的版本不要安装的太高,建议1.10.1
conda create -n 3DLink python=3.9.12
conda install pytorch==1.10.1 torchvision==0.11.2 torchaudio==0.10.1 cudatoolkit=11.3 -c pytorch -c conda-forge
conda install -c rdkit rdkit
pip install docopt
pip install joblib
pip install networkx或者直接使用以下命令进行安装:
conda env create -f env.yml3.使用3Dlinker进行骨架跃迁(使用内置分子案例)
GitHub提供已经训练好的模型,可以直接进行linker生成任务,执行:
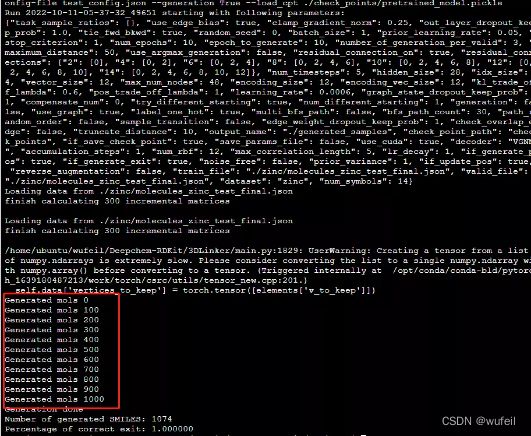
python main.py --dataset zinc --config-file test_config.json --generation True --load_cpt ./check_points/pretrained_model.pickle该命令使用的是./zinc/molecules_zinc_test_final.json作为分子输入文件,是经过处理的,如果需要对自己的分子进行骨架跃迁,请见下一部分。
其中,test_config.json为配置文件,保存输入的分子片段文件,每个分子片段生成分子的数量等信息,内容如下:
{"batch_size": 1, "number_of_generation_per_valid": 10,
"compensate_num": 0, "train_file": "./zinc/molecules_zinc_test_final.json",
"valid_file": "./zinc/molecules_zinc_test_final.json",
"output_name": "./generated_samples" }其中,train_file,valid_file均填入输入的分子片段文件, number_of_generation_per_valid为每个分子生成的分子数量。output_name为生成的分子的保存路径。
执行完毕就会在./generated_samples路径下生成sdf文件和smi文件,其中sdf文件保存了三维的分子构象。文件名分别为:
2022-10-11-05-37-32_49651_generated_smiles_zinc.sdf,
2022-10-11-05-37-32_49651_generated_smiles_zinc.smi.
执行以下命令可以评估生成的分子:
python evaluate_generated_mols.py ZINC PATH_TO_GENERATED_MOLS ../zinc/smi_train.txt 1 True None ./wehi_pains.csv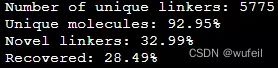

2D filter的通过率为0,这有点奇怪。生成分子的有效率,独特性都正常。
4.使用3Dlinker进行骨架跃迁(使用自己的分子案例)
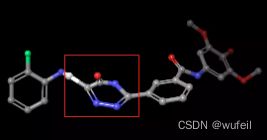
我们的测试案例与Delinker相同:JNK3蛋白的indazole小分子抑制剂,PDB ID为:3FI3,结构如下图所示。
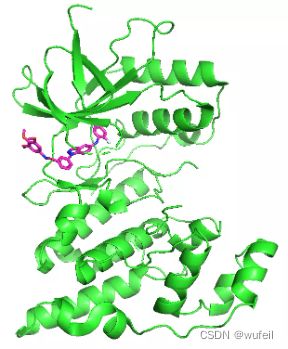
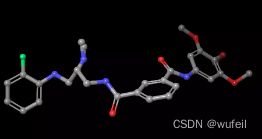
分子的三维构象如下图,保存在./3FI3_ligand.sdf文件内。我们要跃迁的部分如下图红色方框所示的二环,我们称之为骨架跃迁片段,两侧的分子片段称之为片段,或者fragments。

需要注意的是,在数据预处理过程中,并没有直接使用这个构象。因为模型在设计时使用的是ZINC数据集,其中并不包含分子的三维构象信息。所以作者使用RDKIT生成20各分子构象,选择其中能量最低的构象的坐标输入模型。
因此,即使JNK3蛋白的indazole小分子抑制剂已经存在了三维的构象信息,但是还是需要现将分子转化为smi,然后输入到3Dlinker模型之中。
4.1 数据准备
首先,执行路径切换至3DLinker/zinc,新建一个名为Jupyter_Case的文件夹,在其中新建一个,名为:fragments_split_for_3DLinker的ipynb文件。
添加执行路径:
import sys
sys.path.append("../../")
sys.path.append("../../analysis/")导入相关的包:
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit.Chem import Draw
from rdkit.Chem.Draw import IPythonConsole
from rdkit.Chem.Draw import MolDrawing, DrawingOptions
from rdkit.Chem import MolStandardize
import numpy as np
from itertools import product
from joblib import Parallel, delayed
import re
from collections import defaultdict
from IPython.display import clear_output
IPythonConsole.ipython_useSVG = True
import example_utils
import frag_utils加载./3FI3_ligand.sdf,查看一下分子结构:
scaff_1_path = './3FI3_ligand.sdf'
scaff_1_sdf = Chem.SDMolSupplier(scaff_1_path)
scaff_1_smi = Chem.MolToSmiles(scaff_1_sdf[0])
img = Draw.MolsToGridImage([Chem.MolFromSmiles(scaff_1_smi)], molsPerRow=2, subImgSize=(300, 300))
img
计算分子的二维坐标,对原子进行编号:
starting_point_2d = Chem.Mol(scaff_1_sdf[0])
_ = AllChem.Compute2DCoords(starting_point_2d)
example_utils.mol_with_atom_index(starting_point_2d) 
确定要跃迁的部分,这里选择[5,16], [4, 24]键之间的双环部分。现在进行分子切割:
atom_pair_idx_1 = [19, 21]
atom_pair_idx_2 = [5, 16]
bonds_to_break = [starting_point_2d.GetBondBetweenAtoms(x,y).GetIdx() for x,y in [atom_pair_idx_1, atom_pair_idx_2]]
fragmented_mol = Chem.FragmentOnBonds(starting_point_2d, bonds_to_break)
_ = AllChem.Compute2DCoords(fragmented_mol)
fragmented_mol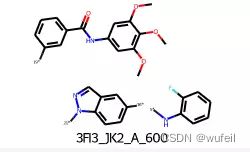
提取骨架片段和分子的smiles:将连接处使用*占位
# Split fragmentation into core and fragments
fragmentation = Chem.MolToSmiles(fragmented_mol).split('.')
fragments = []
for fragment in fragmentation:
if len([x for x in fragment if x =="*"]) ==2:
linker=fragment
else:
fragments.append(fragment)
fragments = '.'.join(fragments)
linker = re.sub('[0-9]+\*', '*', linker)
fragments = re.sub('[0-9]+\*', '*', fragments)
linker, fragments输出:
('[*]c1ccc2c(cnn2[*])c1',
'[*]c1cccc(C(=O)Nc2cc(OC)c(OC)c(OC)c2)c1.[*]Nc1ccccc1F')
将骨架跃迁片段和分子写入txt文件内:
# Write data to file
data_path = "./scaffold_hopping_test_data.txt"
with open(data_path, 'w') as f:
f.write("%s %s" % (fragments, scaff_1_smi))执行完成后,会在当前路径下生成./scaffold_hopping_test_data.txt,用于下一步数据预处理。
接下来使用3DLinker提供的脚本进行预处理,分为两步:
注:需要先将执行路径切换至zinc文件夹内。
第一步,通过raw_preprocessing.py脚本,将数据组装成分子smiles,骨架跃迁片段,分子片段,两个分子片段之间的距离和角度(由于3Dlinker模型不需要输入该数据,raw_preprocessing.py计算距离和角度,而是直接设置为0):
python raw_preprocessing.py --data_path ./Jupyter_Case/scaffold_hopping_test_data.txt --output_path ./Jupyter_Case/scaffold_hopping_test_data.json --verbose--data_path为输入的文件的路径,使用刚才生成的./scaffold_hopping_test_data.txt
--output_path为输出文件的路径,scaffold_hopping_test_data.json
第二步,使用第一步输出的文件,将分子进行图表示:
python prepare_data.py --data_path ./Jupyter_Case/scaffold_hopping_test_data.json --dataset_name ./Jupyter_Case/moleculers_scanffold_hopping输出的文件保存在moleculers_scanffold_hopping.json。
4.2 骨架跃迁
首先,执行路径切换至3DLinker
将数据预处理生成的moleculers_scanffold_hopping.json文件填写到配置文件sanffold_hopping_config.json内,配置完成后的内容如下:
{"batch_size": 1,
"number_of_generation_per_valid": 10,
"compensate_num": 0,
"train_file": "./zinc/Jupyter_Case/moleculers_scanffold_hopping.json", "valid_file": "./zinc/Jupyter_Case/moleculers_scanffold_hopping.json",
"output_name": "./zinc/Jupyter_Case/" }然后执行:
python main.py --dataset zinc --config-file sanffold_hopping_config.json --generation True --load_cpt ./check_points/pretrained_model.picklesanffold_hopping_config.json内容如下:
{"batch_size": 1, "number_of_generation_per_valid": 10, "compensate_num": 0, "train_file": "./zinc/Jupyter_Case/moleculers_scanffold_hopping.json", "valid_file": "./zinc/Jupyter_Case/moleculers_scanffold_hopping.json",
"output_name": "./zinc/Jupyter_Case/" }执行完成后,在3DLinker/zinc/Jupyter_Case目录下生成:
2022-10-11-08-44-20_52851_generated_smiles_zinc.smi和2022-10-11-08-44-20_52851_generated_smiles_zinc.sdf文件,其中sdf文件保存了分子的三维构象信息。
但是生成的sdf分子中的构象,有一些奇怪,有的苯环并不在一个平面内,有的CN键的角度不对。所以,对于该方法的使用还是需要重新进行构象生成。
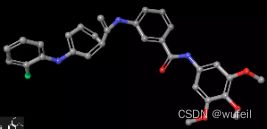
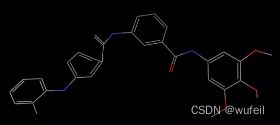
4.3 分子评估
3DLinker的作者提供了进行分子评估的代码,切换到analysis路径下,执行:
python evaluate_generated_mols.py ZINC ../zinc/Jupyter_Case/2022-10-20-08-59-54_19831_generated_smiles_zinc.smi ../zinc/smi_train.txt 1 True None ./wehi_pains.csv注意,要使用smi文件。
输出:
除了使用3DLinker自带的上述脚本进行分子评估,也可以进行如下2D&3D评估。
4.3.1 2D评估
以下代码放置在Jupyter_Case进行。
执行数据预处理和分子骨架跃迁,请参考文档内容,注意执行路径的变化
现在对生成的分子,进行评估,生成的分子保存在:2022-10-20-08-59-54_19831_generated_smiles_zinc.smi。
导入相关的包:
import sys
sys.path.append("../../")
sys.path.append("../../analysis/")
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit.Chem import Draw
from rdkit.Chem.Draw import IPythonConsole
from rdkit.Chem.Draw import MolDrawing, DrawingOptions
from rdkit.Chem import MolStandardize
from IPython.display import clear_output
IPythonConsole.ipython_useSVG = True
from joblib import Parallel, delayed
import re
import csv
import numpy as np
import rdkit_conf_parallel
import frag_utils
import example_utils
import sascorer指定使用的CPU数量:
n_cores = 24加载原始分子
scaff_1_path = './3FI3_ligand.sdf'
scaff_1_sdf = Chem.SDMolSupplier(scaff_1_path)
scaff_1_smi = Chem.MolToSmiles(scaff_1_sdf[0])
img = Draw.MolsToGridImage([Chem.MolFromSmiles(scaff_1_smi)], molsPerRow=1, subImgSize=(300, 300))
img
starting_point_2d = Chem.Mol(scaff_1_sdf[0])
_ = AllChem.Compute2DCoords(starting_point_2d)
example_utils.mol_with_atom_index(starting_point_2d)
按照之前的切割方式,生成骨架跃迁片段和fragments。
atom_pair_idx_1 = [19, 21]
atom_pair_idx_2 = [5, 16]
bonds_to_break = [starting_point_2d.GetBondBetweenAtoms(x,y).GetIdx() for x,y in [atom_pair_idx_1, atom_pair_idx_2]]
fragmented_mol = Chem.FragmentOnBonds(starting_point_2d, bonds_to_break)
_ = AllChem.Compute2DCoords(fragmented_mol)
fragmented_mol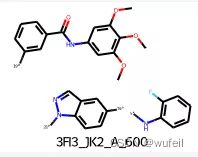
fragmentation = Chem.MolToSmiles(fragmented_mol).split('.')
fragments = []
for fragment in fragmentation:
if len([x for x in fragment if x =="*"]) ==2:
linker=fragment
else:
fragments.append(fragment)
fragments = '.'.join(fragments)
linker = re.sub('[0-9]+\*', '*', linker)
fragments = re.sub('[0-9]+\*', '*', fragments)
fragments输出:'[*]c1cccc(C(=O)Nc2cc(OC)c(OC)c(OC)c2)c1.[*]Nc1ccccc1F'
对骨架跃迁生成的分子进行初步处理
generated_smiles = frag_utils.read_triples_file("./2022-10-20-08-59-54_19831_generated_smiles_zinc.smi")
in_mols = [smi[1] for smi in generated_smiles]
frag_mols = [smi[0] for smi in generated_smiles]
gen_mols = [smi[2] for smi in generated_smiles]
du = Chem.MolFromSmiles('*')
clean_frags = [Chem.MolToSmiles(Chem.RemoveHs(AllChem.ReplaceSubstructs(Chem.MolFromSmiles(smi),du,Chem.MolFromSmiles('[H]'),True)[0])) for smi in frag_mols]
clear_output(wait=True)
print("Done")检查生成分子的有效性
results = []
for in_mol, frag_mol, gen_mol, clean_frag in zip(in_mols, frag_mols, gen_mols, clean_frags):
if len(Chem.MolFromSmiles(gen_mol).GetSubstructMatch(Chem.MolFromSmiles(clean_frag)))>0:
results.append([in_mol, frag_mol, gen_mol, clean_frag])
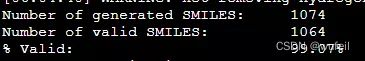
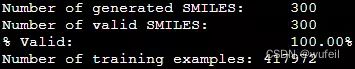
print("Number of generated SMILES: \t%d" % len(generated_smiles))
print("Number of valid SMILES: \t%d" % len(results))
print("%% Valid: \t\t\t%.2f%%" % (len(results)/len(generated_smiles)*100))输出:
Number of generated SMILES: 300
Number of valid SMILES: 300
% Valid: 100.00%分子有效性100%,还不错。
找出生成分子的linker,即骨架跃迁部分
linkers = Parallel(n_jobs=n_cores)(delayed(frag_utils.get_linker)(Chem.MolFromSmiles(m[2]), Chem.MolFromSmiles(m[3]), m[1]) \
for m in results)
for i, linker in enumerate(linkers):
if linker == "":
continue
linker = Chem.MolFromSmiles(re.sub('[0-9]+\*', '*', linker))
Chem.rdmolops.RemoveStereochemistry(linker)
linkers[i] = MolStandardize.canonicalize_tautomer_smiles(Chem.MolToSmiles(linker))
for i in range(len(results)):
results[i].append(linkers[i])
clear_output(wait=True)创建所有骨架跃迁分子的目录,并输出生成分子的unique比例
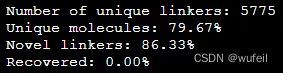
results_dict = {}
for res in results:
if res[0]+'.'+res[1] in results_dict: # Unique identifier - starting fragments and original molecule
results_dict[res[0]+'.'+res[1]].append(tuple(res))
else:
results_dict[res[0]+'.'+res[1]] = [tuple(res)]
print("Unique molecules: %.2f%%" % (frag_utils.unique(results_dict.values())*100))Unique molecules: 79.67%,说明生成分子的重复率有点高。
检查骨架跃迁分子通过2Dfilter的情况:
filters_2d = frag_utils.calc_filters_2d_dataset(results, pains_smarts_loc="../../analysis/wehi_pains.csv", n_cores=n_cores)
results_filt = []
for res, filt in zip(results, filters_2d):
if filt[0] and filt[1] and filt[2]:
results_filt.append(res)
clear_output(wait=True)
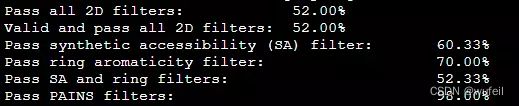
print("Pass all 2D filters: \t\t\t\t%.2f%%" % (len(results_filt)/len(results)*100))
print("Valid and pass all 2D filters: \t\t\t%.2f%%" % (len(results_filt)/len(generated_smiles)*100))
print("Pass synthetic accessibility (SA) filter: \t%.2f%%" % (len([f for f in filters_2d if f[0]])/len(filters_2d)*100))
print("Pass ring aromaticity filter: \t\t\t%.2f%%" % (len([f for f in filters_2d if f[1]])/len(filters_2d)*100))
print("Pass SA and ring filters: \t\t\t%.2f%%" % (len([f for f in filters_2d if f[0] and f[1]])/len(filters_2d)*100))
print("Pass PAINS filters: \t\t\t\t%.2f%%" % (len([f for f in filters_2d if f[2]])/len(filters_2d)*100))输出:
Pass all 2D filters: 52.00%
Valid and pass all 2D filters: 52.00%
Pass synthetic accessibility (SA) filter: 60.33%
Pass ring aromaticity filter: 70.00%
Pass SA and ring filters: 52.33%
Pass PAINS filters: 98.00%
print("Number molecules passing 2D filters:\t\t%d" % len(results_filt))
results_filt_unique = example_utils.unique_mols(results_filt)
print("Number unique molecules passing 2D filters:\t%d" % len(results_filt_unique))输出:
Number molecules passing 2D filters: 156
Number unique molecules passing 2D filters: 114
4.3.2 3D评估
接下来进行三维形状的评估。
首先为每一个分子生成三维构象。
_ = rdkit_conf_parallel.gen_confs([res[2] for res in results_filt], "./3DLinker_generated_mols_unique.sdf",
smi_frags=[res[1] for res in results_filt], numcores=n_cores, jpsettings=True)
clear_output(wait=True)
print("Done")加载生成的构象:
gen_sdfs = Chem.SDMolSupplier("./3DLinker_generated_mols_unique.sdf")
ref_mol = Chem.Mol(scaff_1_sdf[0])获得片段的构象及其编号
used = set([])
ref_identifiers = [(res[1], res[0]) for res in results_filt if res[1]+'.'+res[0] not in used and (used.add(res[1]+'.'+res[0]) or True)]
# Get indices of compounds in SD file
start_stop_idxs = []
start = 0
errors = 0
curr_st_pt = ""
for count, gen_mol in enumerate(gen_sdfs):
try:
# Check if seen this ligand before
if gen_mol.GetProp("_Model") == str(0):
stop = count
if count != 0:
start_stop_idxs.append((start, stop))
start = int(stop) # deep copy
curr_st_pt = gen_mol.GetProp("_StartingPoint")
except:
errors += 1
continue
# Add last
start_stop_idxs.append((start, len(gen_sdfs)))计算SC_RDKit片段分数
# Calculate SC_RDKit fragments scores
names_frags = []
best_scores_frags = []
idx_best_poses_frags = []
names_frags_start_pts = []
with Parallel(n_jobs=n_cores, backend='multiprocessing') as parallel:
for i in range(-(-len(start_stop_idxs)//n_cores)):
jobs = []
for core in range(n_cores):
if i*n_cores+core < len(start_stop_idxs):
start, stop = start_stop_idxs[i*n_cores+core]
frag_smi = gen_sdfs[start].GetProp("_StartingPoint")
# Prepare jobs
gen_mols = [(Chem.Mol(gen_sdfs[idx]), Chem.Mol(ref_mol), str(frag_smi)) for idx in range(start, stop) if gen_sdfs[idx] is not None] # Test addition
jobs.append(gen_mols)
# Get SC_RDKit scores
set_scores = parallel((delayed(frag_utils.SC_RDKit_frag_scores)(gen_mols) for gen_mols in jobs))
for core, scores in enumerate(set_scores):
start, stop = start_stop_idxs[i*n_cores+core]
names_frags.append(gen_sdfs[start].GetProp("_Name"))
names_frags_start_pts.append(gen_sdfs[start].GetProp("_StartingPoint"))
best_scores_frags.append(max(scores))
idx_best_poses_frags.append((np.argmax(scores)+start, 0))
best_scores_frags_all = []
comp = list(zip(names_frags_start_pts, names_frags))
for res in results:
try:
idx = comp.index((res[1], res[2]))
best_scores_frags_all.append(best_scores_frags[idx])
except:
continue输出评估结果
print("Average SC_RDKit Fragments score: %.3f +- %.3f\n" % (np.mean(best_scores_frags_all), np.std(best_scores_frags_all)))
thresholds_SC_RDKit = [0.6, 0.7, 0.75, 0.8]
for thresh in thresholds_SC_RDKit:
print("SC_RDKit Fragments - Molecules above %.2f: %.2f%%" % (thresh, len([score for score in best_scores_frags_all if score >= thresh]) / len(best_scores_frags_all)*100))输出:
Average SC_RDKit Fragments score: 0.716 +- 0.044
SC_RDKit Fragments - Molecules above 0.60: 100.00%
SC_RDKit Fragments - Molecules above 0.70: 61.54%
SC_RDKit Fragments - Molecules above 0.75: 21.79%
SC_RDKit Fragments - Molecules above 0.80: 5.77%
计算骨架跃迁生成的分子和原来分子的RMSD
names_rmsd_frags = []
best_rmsd_frags = []
idx_best_rmsd_poses_frags = []
names_rmsd_frags_start_pts = []
with Parallel(n_jobs=n_cores, backend='multiprocessing') as parallel:
for i in range(-(-len(start_stop_idxs)//n_cores)):
jobs = []
for core in range(n_cores):
if i*n_cores+core < len(start_stop_idxs):
start, stop = start_stop_idxs[i*n_cores+core]
frag_smi = gen_sdfs[start].GetProp("_StartingPoint")
# Prepare jobs
gen_mols = [(Chem.Mol(gen_sdfs[idx]), Chem.Mol(ref_mol), str(frag_smi)) for idx in range(start, stop) if gen_sdfs[idx] is not None] # Test addition
jobs.append(gen_mols)
# Calculate RMSDs
set_scores = parallel((delayed(frag_utils.rmsd_frag_scores)(gen_mols) for gen_mols in jobs)) # Multiprocessing step
for core, scores in enumerate(set_scores):
start, stop = start_stop_idxs[i*n_cores+core]
names_rmsd_frags.append(gen_sdfs[start].GetProp("_Name"))
names_rmsd_frags_start_pts.append(gen_sdfs[start].GetProp("_StartingPoint"))
best_rmsd_frags.append(min(scores))
idx_best_rmsd_poses_frags.append((np.argmin(scores)+start, 0))
best_rmsd_frags_all = []
comp = list(zip(names_rmsd_frags_start_pts, names_rmsd_frags))
for res in results:
try:
idx = comp.index((res[1], res[2]))
best_rmsd_frags_all.append(best_rmsd_frags[idx])
except:
continue输出RMSD的评估结果:
print("Average Fragments RMSD: %.3f +- %.3f\n" % (np.mean(best_rmsd_frags_all), np.std(best_rmsd_frags_all)))
thresholds_rmsd = [1.0, 0.75, 0.5]
for thresh in thresholds_rmsd:
print("RMSD Fragments - Molecules below %.2f: %.2f%%" % (thresh, len([score for score in best_rmsd_frags_all if score <= thresh]) / len(best_rmsd_frags_all)*100))Average Fragments RMSD: 0.984 +- 0.160
RMSD Fragments - Molecules below 1.00: 57.05%
RMSD Fragments - Molecules below 0.75: 6.41%
RMSD Fragments - Molecules below 0.50: 0.00%
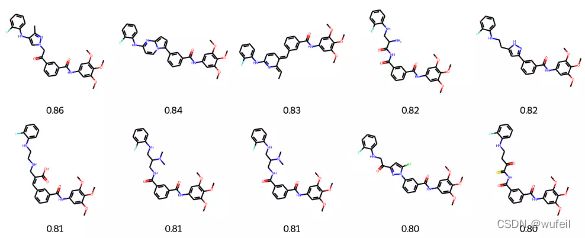
4.3.4 Top10 最佳结果
输出最佳的SC_RDKit 打分结果:
best_mols = sorted(list(zip(names_frags, best_scores_frags)), key=lambda x: x[1], reverse=True)[:10]
mols = [Chem.MolFromSmiles(m[0]) for m in best_mols]
frag_to_align = re.sub('\[\*\]', '', fragments.split('.')[0])
p = Chem.MolFromSmiles(frag_to_align)
AllChem.Compute2DCoords(p)
for m in mols: AllChem.GenerateDepictionMatching2DStructure(m,p)
Draw.MolsToGridImage(mols,
molsPerRow=5, legends=["%.2f" % m[1] for m in best_mols]
)
输出RMSD最佳的结果
best_mols = sorted(list(zip(names_rmsd_frags, best_rmsd_frags)), key=lambda x: x[1])[:10]
mols = [Chem.MolFromSmiles(m[0]) for m in best_mols]
frag_to_align = re.sub('\[\*\]', '', fragments.split('.')[0])
p = Chem.MolFromSmiles(frag_to_align)
AllChem.Compute2DCoords(p)
for m in mols: AllChem.GenerateDepictionMatching2DStructure(m,p)
Draw.MolsToGridImage(mols,
molsPerRow=5, legends=["%.2f" % m[1] for m in best_mols]
)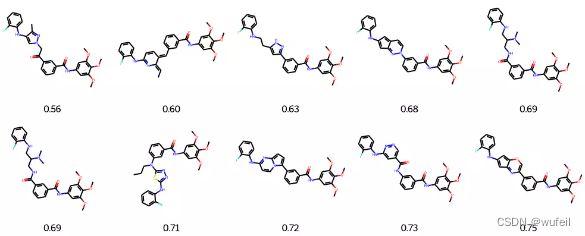
这一结果与之前的Delinker相比,SC_RDKit分数提高了,同时分子的RMSD有所降低,说明该方法要比Delinker更好一些。
注:github中的源代码存在文件缺失,代码需要修改,才能复现上述结果。